Structural and Dynamic Characterizations Highlight the Deleterious Role of SULT1A1 R213H Polymorphism in Substrate Binding

 ,
,  ,
, 
 and
and 
Abstract
:
1. Introduction
2. Results
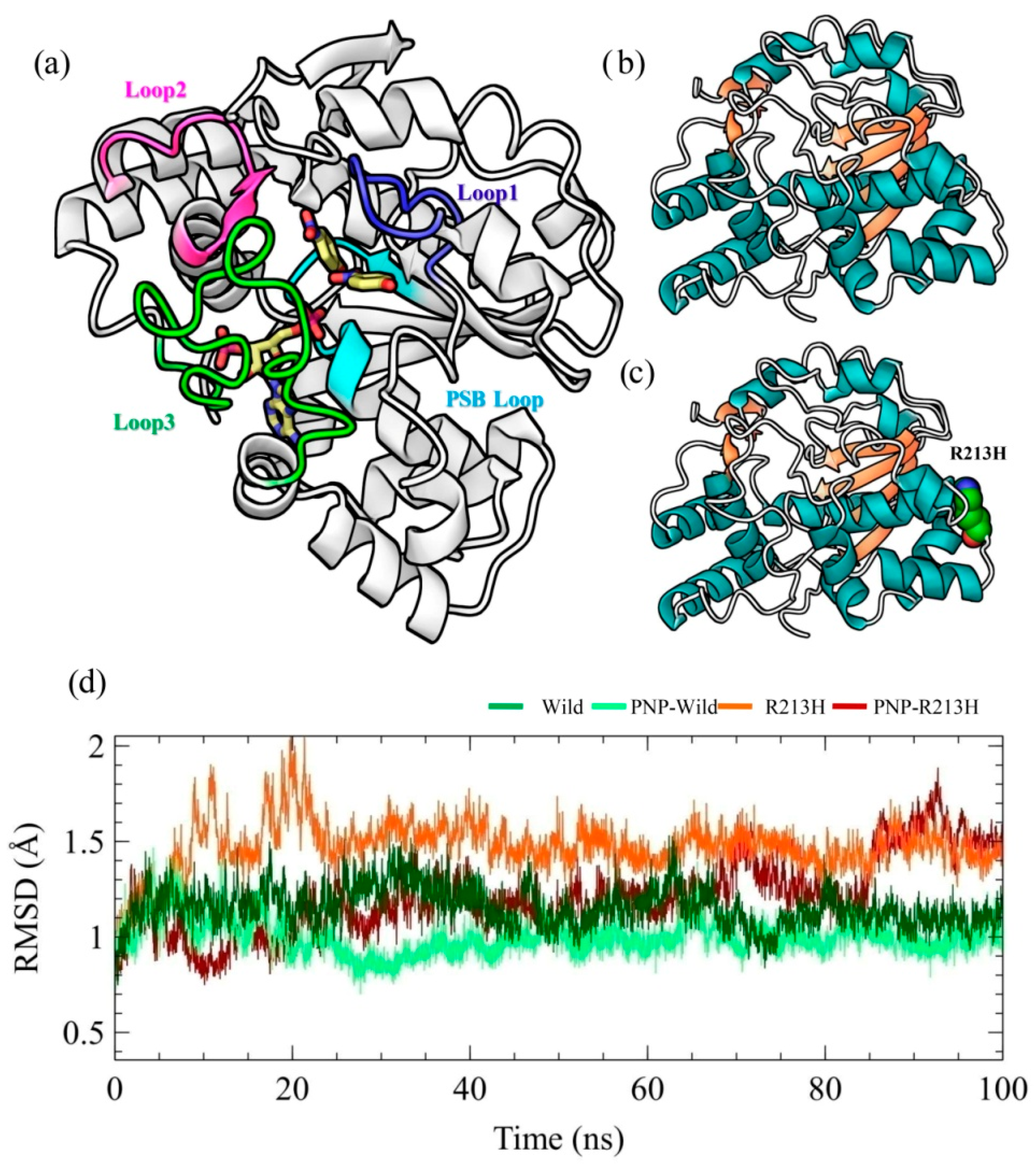
2.1. Architecture of SULT1A1 and Stability of Simulation Systems
2.2. Effects of Mutation on Conformation Stability
2.3. Effects of Mutation in Protein Dynamics
2.4. Effects of Mutation in Active Site and Ligand Binding
2.5. Insights Into Substrate Binding
3. Discussion
4. Materials and Methods
4.1. Preparation of the Simulation System
4.2. Molecular Dynamics Simulation
4.3. Dynamic Cross-Correlation Map (DCCM) Analysis
4.4. Principal Component Analysis (PCA)
4.5. Free Energy Landscape Analysis
4.6. Per Residue Energy Decomposition Analysis
ΔGbind = ΔEMM + ΔGsol − TΔS (e2),
ΔEMM = ΔEelec + ΔEvdW (e3),
ΔGsol = ΔGpol + ΔGnpol (e4),
ΔGnpol = γ × SASA + b (e5),
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Huang, X.; Cao, M.; Wang, L.; Wu, S.; Liu, X.; Li, H.; Zhang, H.; Wang, R.Y.; Sun, X.; Wei, C.; et al. Expression of sulfotransferase SULT1A1 in cancer cells predicts susceptibility to the novel anticancer agent NSC-743380. Oncotarget 2015, 6, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Gamage, N.; Barnett, A.; Hempel, N.; Duggleby, R.G.; Windmill, K.F.; Martin, J.L.; McManus, M.E. Human sulfotransferases and their role in chemical metabolism. Toxicol. Sci. 2005, 90, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Glatt, H. Sulfotransferases in the bioactivation of xenobiotics. Chem. Biol. Inter. 2000, 129, 141–170. [Google Scholar] [CrossRef]
- Glatt, H.; Boeing, H.; Engelke, C.E.; Ma, L.; Kuhlow, A.; Pabel, U.; Pomplun, D.; Teubner, W.; Meinl, W. Human cytosolic sulphotransferases: Genetics, characteristics, toxicological aspects. Mutat. Res. 2001, 482, 27–40. [Google Scholar] [CrossRef]
- Barnett, A.C.; Tsvetanov, S.; Gamage, N.; Martin, J.L.; Duggleby, R.G.; McManus, M.E. Active site mutations and substrate inhibition in human sulfotransferase 1A1 and 1A3. J. Biol. Chem. 2004, 279, 18799–18805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, I.; Guttman, C.; Amar, D.; Zarivach, R.; Aharoni, A. The molecular basis for the broad substrate specificity of human sulfotransferase 1A1. PLoS ONE 2011, 6, e26794. [Google Scholar] [CrossRef] [Green Version]
- Pachouri, S.S.; Sobti, R.C.; Kaur, P.; Singh, J.; Gupta, S. Impact of polymorphism in sulfotransferase gene on the risk of lung cancer. Cancer Genet. Cytogenet. 2006, 171, 39–43. [Google Scholar] [CrossRef]
- Li, K.; Ren, Y.W.; Wan, Y.; Yin, Z.H.; Wu, W.; Zhou, B.S. SULT1A1 Arg213His polymorphism and susceptibility of environment-related cancers: A meta analysis of 5915 cases and 7900 controls. Mol. Biol. Rep. 2012, 39, 2597–2605. [Google Scholar] [CrossRef]
- Glatt, H. Sulfation and sulfotransferases 4: Bioactivation of mutagens via sulfation. FASEB J. 1997, 11, 314–321. [Google Scholar] [CrossRef]
- Weinshilboum, R.M.; Otterness, D.M.; Aksoy, I.A.; Wood, T.C.; Her, C.; Raftogianis, R.B. Sulfation and sulfotransferases 1: Sulfotransferase molecular biology: cDNAs and genes. FASEB J. 1997, 11, 3–14. [Google Scholar] [CrossRef]
- Liang, G.; Miao, X.; Zhou, Y.; Tan, W.; Lin, D. A functional polymorphism in the SULT1A1 gene (G638A) is associated with risk of lung cancer in relation to tobacco smoking. Carcinogenesis 2004, 25, 773–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, H.; Lang, N.; Kadlubar, F. Metabolic activation of N-hydroxyarylamines and N-hydroxy heterocyclic amines by human sulfotransferase (s). Cancer Res. 1995, 55, 525–529. [Google Scholar] [PubMed]
- Yamazoe, Y.; Nagata, K.; Yoshinari, K.; Fujita, K.; Shiraga, T.; Iwasaki, K. Sulfotransferase catalyzing sulfation of heterocyclic amines. Cancer Lett. 1999, 143, 103–107. [Google Scholar] [CrossRef]
- Banoglu, E. Current status of the cytosolic sulfotransferases in the metabolic activation of promutagens and procarcinogens. Curr. Drug Metab. 2000, 1, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmagadda, D.; Cherala, G.; Ghatta, S. Cytosolic sulfotransferases. Indian J. Exp. Biol. 2006, 44, 171–182. [Google Scholar]
- Wang, Z.; Fu, Y.; Tang, C.; Lu, S.; Chu, W.-m. SULT1A1 R213H polymorphism and breast cancer risk: A meta-analysis based on 8454 cases and 11,800 controls. Breast Cancer Res. Treat. 2010, 122, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Tasnim, T.; Al-Mamun, M.M.A.; Nahid, N.A.; Islam, M.R.; Apu, M.N.H.; Bushra, M.U.; Rabbi, S.N.I.; Nahar, Z.; Chowdhury, J.A.; Ahmed, M.U. Genetic variants of SULT1A1 and XRCC1 genes and risk of lung cancer in Bangladeshi population. Tumor Biol. 2017, 39, 1010428317729270. [Google Scholar] [CrossRef] [Green Version]
- Forat-Yazdi, M.; Jafari, M.; Kargar, S.; Abolbaghaei, S.M.; Nasiri, R.; Farahnak, S.; Foroughi, E.; Neamatzadeh, H. Association between SULT1A1 Arg213His (Rs9282861) polymorphism and risk of breast cancer: A systematic review and meta-analysis. J. Res. Health Sci. 2017, 17, e00396. [Google Scholar]
- Jiang, Y.; Zhou, L.; Yan, T.; Shen, Z.; Shao, Z.; Lu, J. Association of sulfotransferase SULT1A1 with breast cancer risk: A meta-analysis of case-control studies with subgroups of ethnic and menopausal statue. J. Exp. Clin. Cancer Res. 2010, 29, 101. [Google Scholar] [CrossRef] [Green Version]
- Francis, A.M.; Ramya, R.; Ganesan, N.; Kumarasamy, P.; Paul, S.F.; Munirajan, A.; Divya, M. Breast cancer susceptibility genes in estrogen metabolizing pathway in a southern Indian population. Meta Gene 2019, 19, 225–234. [Google Scholar] [CrossRef]
- Li, W.; Gu, M. SULT1A1 Arg213His polymorphism is associated with bladder cancer risk: A meta-analysis. Med. Sci. Monit. 2014, 20, 1590. [Google Scholar] [PubMed] [Green Version]
- Boccia, S.; Persiani, R.; La Torre, G.; Rausei, S.; Arzani, D.; Gianfagna, F.; Romano-Spica, V.; D’Ugo, D.; Ricciardi, G. Sulfotransferase 1A1 polymorphism and gastric cancer risk: A pilot case-control study. Cancer Lett. 2005, 229, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Boccia, S.; Cadoni, G.; La Torre, G.; Arzani, D.; Volante, M.; Cattel, C.; Gianfagna, F.; Paludetti, G.; Almadori, G.; Ricciardi, G. A case–control study investigating the role of sulfotransferase 1A1 polymorphism in head and neck cancer. J. Cancer Res. Clin. Oncol. 2006, 132, 466. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Spitz, M.R.; Tsou, A.M.-H.; Zhang, K.; Makan, N.; Wu, X. Sulfotransferase (SULT) 1A1 polymorphism as a predisposition factor for lung cancer: A case-control analysis. Lung Cancer 2002, 35, 137–142. [Google Scholar] [CrossRef]
- Lopes, B.A.; Emerenciano, M.; Gonçalves, B.A.A.; Vieira, T.M.; Rossini, A.; Pombo-de-Oliveira, M.S. Polymorphisms in CYP1B1, CYP3A5, GSTT1, and SULT1A1 are associated with early age acute leukemia. PLoS ONE 2015, 10, e0127308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, K.; Yamazoe, Y. Pharmacogenetics of sulfotransferase. Ann. Rev. Pharmacol. Toxicol. 2000, 40, 159–176. [Google Scholar] [CrossRef]
- Nagar, S.; Walther, S.; Blanchard, R.L. Sulfotransferase (SULT) 1A1 polymorphic variants* 1,* 2, and* 3 are associated with altered enzymatic activity, cellular phenotype, and protein degradation. Mol. Pharmacol. 2006, 69, 2084–2092. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, A.; Ferrari, A.; Ottani, A.; Guerzoni, S.; Tacchi, R.; Leone, S. Paracetamol: New vistas of an old drug. CNS Drug Rev. 2006, 12, 250–275. [Google Scholar] [CrossRef]
- Rasool, M.I.; Bairam, A.F.; Gohal, S.A.; El Daibani, A.A.; Alherz, F.A.; Abunnaja, M.S.; Alatwi, E.S.; Kurogi, K.; Liu, M.-C. Effects of the human SULT1A1 polymorphisms on the sulfation of acetaminophen, O-desmethylnaproxen, and tapentadol. Pharmacol. Rep. 2019, 71, 257–265. [Google Scholar] [CrossRef]
- Wang, L.-Q.; James, M.O. Inhibition of sulfotransferases by xenobiotics. Curr. Drug Metab. 2006, 7, 83–104. [Google Scholar] [CrossRef]
- Gamage, N.U.; Duggleby, R.G.; Barnett, A.C.; Tresillian, M.; Latham, C.F.; Liyou, N.E.; McManus, M.E.; Martin, J.L. Structure of a human carcinogen-converting enzyme, SULT1A1 Structural and kinetic implications of substrate inhibition. J. Biol. Chem. 2003, 278, 7655–7662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidwell, L.M.; McManus, M.E.; Gaedigk, A.; Kakuta, Y.; Negishi, M.; Pedersen, L.; Martin, J.L. Crystal structure of human catecholamine sulfotransferase. J. Mol. Boil. 1999, 293, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, S.; Petrotchenko, E.V.; Pedersen, L.C.; Negishi, M. Crystallographic analysis of a hydroxylated polychlorinated biphenyl (OH-PCB) bound to the catalytic estrogen binding site of human estrogen sulfotransferase. Environ. Health Perspect. 2003, 111, 884–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Rabjohn, P.A.; York, J.L.; Wooldridge, C.; Zhang, D.; Falany, C.N.; Radominska-Pandya, A. Carboxyl residues in the active site of human phenol sulfotransferase (SULT1A1). Biochemistry 2000, 39, 16000–16007. [Google Scholar] [CrossRef] [PubMed]
- Duggleby, R.G.; Gaedigk, A.; Mcmanus, M.E. Structural characterization of human aryl sulphotransferases. Biochem. J. 1999, 337, 337–343. [Google Scholar]
- Dong, D.; Ako, R.; Wu, B. Crystal structures of human sulfotransferases: Insights into the mechanisms of action and substrate selectivity. Expert Opin. Drug Metabol. Toxicol. 2012, 8, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Thirumal, D.K.; Iyer, S.; Christy, J.P.; Siva, R.; Tayubi, I.A.; George, C.P.D.; Zayed, H. A comparative computational approach toward pharmacological chaperones (NN-DNJ and ambroxol) on N370S and L444P mutations causing Gaucher’s disease. Adv. Protein Chem. Struct. Biol. 2019, 114, 315–339. [Google Scholar]
- Hannan, M.; Dash, R.; Sohag, A.A.M.; Moon, I.S. Deciphering Molecular Mechanism of the Neuropharmacological Action of Fucosterol through Integrated System Pharmacology and InSilico Analysis. Mar. Drugs 2019, 17, 639. [Google Scholar] [CrossRef] [Green Version]
- Thirumal, D.K.; Judith, E.; Priyadharshini, J.C.; Siva, R.; Tayubi, I.A.; Chakraborty, C.; George, C.P.D.; Zayed, H. Computational and modeling approaches to understand the impact of the Fabry’s disease causing mutation (D92Y) on the interaction with pharmacological chaperone 1-deoxygalactonojirimycin (DGJ). Adv. Protein Chem. Struct. Biol. 2019, 114, 341–407. [Google Scholar]
- Kumar, D.T.; Emerald, L.J.; Doss, C.G.P.; Sneha, P.; Siva, R.; Jebaraj, W.C.E.; Zayed, H. Computational approach to unravel the impact of missense mutations of proteins (D2HGDH and IDH2) causing D-2-hydroxyglutaric aciduria 2. Metab. Brain Dis. 2018, 33, 1699–1710. [Google Scholar] [CrossRef]
- Tang, X.; Wang, Z.; Lei, T.; Zhou, W.; Chang, S.; Li, D. Importance of protein flexibility on molecular recognition: Modeling binding mechanisms of aminopyrazine inhibitors to Nek2. Phys. Chem. Chem. Phys. 2018, 20, 5591–5605. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.; Dong, Z. Concerted dynamic motions of an FABP4 model and its ligands revealed by microsecond molecular dynamics simulations. Biochemistry 2014, 53, 6409–6417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodsdon, M.E.; Cistola, D.P. Ligand binding alters the backbone mobility of intestinal fatty acid-binding protein as monitored by 15N NMR relaxation and 1H exchange. Biochemistry 1997, 36, 2278–2290. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Lin, C.L.; Tang, C.; Ponder, J.W.; Kao, J.L.; Cistola, D.P.; Li, E. Binding of retinol induces changes in rat cellular retinol-binding protein II conformation and backbone dynamics. J. Mol. Boil. 2000, 300, 619–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzoni, L.; Lucke, C.; Perez, C.; Cavazzini, D.; Rademacher, M.; Ludwig, C.; Spisni, A.; Rossi, G.L.; Ruterjans, H. Structure and backbone dynamics of Apo- and holo-cellular retinol-binding protein in solution. J. Boil. Chem. 2002, 277, 21983–21997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, J.M.; Amat, M.; Morgan, B.R.; Royer, W.E., Jr.; Massi, F. Insight into the allosteric mechanism of Scapharca dimeric hemoglobin. Biochemistry 2014, 53, 7199–7210. [Google Scholar] [CrossRef] [Green Version]
- Nemaysh, V.; Luthra, P.M. Computational analysis revealing that K634 and T681 mutations modulate the 3D-structure of PDGFR-β and lead to sunitinib resistance. RSC Adv. 2017, 7, 37612–37626. [Google Scholar] [CrossRef] [Green Version]
- Kamaraj, B.; Purohit, R. In silico screening and molecular dynamics simulation of disease-associated nsSNP in TYRP1 gene and its structural consequences in OCA3. Biomed. Res. Int. 2013, 2013, 697051. [Google Scholar] [CrossRef] [Green Version]
- Kitao, A.; Hirata, F.; Gō, N. The effects of solvent on the conformation and the collective motions of protein: Normal mode analysis and molecular dynamics simulations of melittin in water and in vacuum. Chem. Phys. 1991, 158, 447–472. [Google Scholar] [CrossRef]
- Amadei, A.; Linssen, A.B.; Berendsen, H.J. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Kitao, A.; Go, N. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Boil. 1999, 9, 164–169. [Google Scholar] [CrossRef]
- García, A.E. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 1992, 68, 2696. [Google Scholar] [CrossRef] [PubMed]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Principal component analysis for protein folding dynamics. J. Mol. Boil. 2009, 385, 312–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Relation between free energy landscapes of proteins and dynamics. J. Chem. Theory Comput. 2010, 6, 583–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grottesi, A.; Domene, C.; Hall, B.; Sansom, M.S. Conformational dynamics of M2 helices in KirBac channels: Helix flexibility in relation to gating via molecular dynamics simulations. Biochemistry 2005, 44, 14586–14594. [Google Scholar] [CrossRef]
- Yang, L.; Song, G.; Carriquiry, A.; Jernigan, R.L. Close correspondence between the motions from principal component analysis of multiple HIV-1 protease structures and elastic network modes. Structure 2008, 16, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Lou, H.; Cukier, R.I. Molecular dynamics of apo-adenylate kinase: A principal component analysis. J. Phys. Chem. B 2006, 110, 12796–12808. [Google Scholar] [CrossRef]
- Spellmon, N.; Sun, X.; Sirinupong, N.; Edwards, B.; Li, C.; Yang, Z. Molecular dynamics simulation reveals correlated inter-lobe motion in protein lysine methyltransferase SMYD2. PLoS ONE 2015, 10, e0145758. [Google Scholar] [CrossRef]
- Hosen, S.Z.; Dash, R.; Junaid, M.; Mitra, S.; Absar, N. Identification and structural characterization of deleterious non-synonymous single nucleotide polymorphisms in the human SKP2 gene. Comput. Boil. Chem. 2019, 79, 127–136. [Google Scholar] [CrossRef]
- Agrahari, A.K.; George, P.D.C.; Siva, R.; Magesh, R.; Zayed, H. Molecular insights of the G2019S substitution in LRRK2 kinase domain associated with Parkinson’s disease: A molecular dynamics simulation approach. J. Theor. Boil. 2019, 469, 163–171. [Google Scholar] [CrossRef]
- Kim, H.J.; Choi, M.Y.; Kim, H.J.; Llinás, M. Conformational dynamics and ligand binding in the multi-domain protein PDC109. PLoS ONE 2010, 5, e9180. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Junaid, M.; Mitra, S.; Arifuzzaman, M.; Hosen, S.Z. Structure-based identification of potent VEGFR-2 inhibitors from in vivo metabolites of a herbal ingredient. J. Mol. Model. 2019, 25, 98. [Google Scholar] [CrossRef] [PubMed]
- Raftogianis, R.B.; Wood, T.C.; Weinshilboum, R.M. Human phenol sulfotransferases SULT1A2 and SULT1A1: Genetic polymorphisms, allozyme properties, and human liver genotype–phenotype correlations. Biochem. Pharmacol. 1999, 58, 605–616. [Google Scholar] [CrossRef]
- Raftogianis, R.B.; Wood, T.C.; Otterness, D.M.; Van Loon, J.A.; Weinshilboum, R.M. Phenol sulfotransferase pharmacogenetics in humans: Association of commonSULT1A1alleles with TS PST phenotype. Biochem. Biophys. Res. Commun. 1997, 239, 298–304. [Google Scholar] [CrossRef]
- Ketterer, B.; Mulder, G.J. Glutathione conjugation. In Conjugation Reactions in Drug Metabolism: An Integrated Approach; Taylor & Francis London: London, UK, 1990; pp. 307–364. [Google Scholar]
- Coughtrie, M. Sulfation through the looking glass—recent advances in sulfotransferase research for the curious. Pharmacogn. J. 2002, 2, 297. [Google Scholar] [CrossRef] [Green Version]
- Strott, C.A. Sulfonation and molecular action. Endocr. Rev. 2002, 23, 703–732. [Google Scholar] [CrossRef]
- Shimada, T. Xenobiotic-metabolizing enzymes involved in activation and detoxification of carcinogenic polycyclic aromatic hydrocarbons. Drug Metab. Pharmacokinet. 2006, 21, 257–276. [Google Scholar] [CrossRef] [Green Version]
- Hebbring, S.J.; Adjei, A.A.; Baer, J.L.; Jenkins, G.D.; Zhang, J.; Cunningham, J.M.; Schaid, D.J.; Weinshilboum, R.M.; Thibodeau, S.N. Human SULT1A1 gene: Copy number differences and functional implications. Hum. Mol. Genet. 2006, 16, 463–470. [Google Scholar] [CrossRef]
- Hildebrandt, M.; Carrington, D.; Thomae, B.; Eckloff, B.; Schaid, D.J.; Yee, V.; Weinshilboum, R.M.; Wieben, E.D. Genetic diversity and function in the human cytosolic sulfotransferases. Pharmacogn. J. 2007, 7, 133. [Google Scholar] [CrossRef]
- Kormos, B.L.; Baranger, A.M.; Beveridge, D.L. Do collective atomic fluctuations account for cooperative effects? Molecular dynamics studies of the U1A− RNA complex. J. Am. Chem. Soc. 2006, 128, 8992–8993. [Google Scholar] [CrossRef] [Green Version]
- Hempel, N.; Gamage, N.; Martin, J.L.; McManus, M.E. Human cytosolic sulfotransferase SULT1A1. Int. J. Biochem. Cell Boil. 2007, 39, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, S.; Shimizu, M.; Katoh, T.; Miyajima, A.; Ohno, Y.; Matsumoto, Y.; Fukuoka, M.; Tang, Y.M.; Lang, N.P.; Kadlubar, F.F. Sulfating-activity and stability of cDNA-expressed allozymes of human phenol sulfotransferase, ST1A3*1 ((213)Arg) and ST1A3*2 ((213)His), both of which exist in Japanese as well as Caucasians. J. Biochem. 1999, 126, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.-Y.; Chiang, H.-P.; Chen, W.-T.; Yang, Y.-S. Dimerization is responsible for the structural stability of human sulfotransferase 1A1. Drug Metab. Dispos. 2009, 37, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlić, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2010, 39, D392–D401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, R.; Junaid, M.; Islam, N.; Akash, C.; Forhad, M.; Khan, M.; Arifuzzaman, M.; Khatun, M.; Zahid Hosen, S.M. Molecular insight and binding pattern analysis of Shikonin as a potential VEGFR-2 inhibitor. Curr. Enzyme Inhib. 2017, 13, 235–244. [Google Scholar] [CrossRef]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins Struct. Funct. Bioinform. 2004, 57, 678–683. [Google Scholar] [CrossRef]
- Land, H.; Humble, M.S. YASARA: A tool to obtain structural guidance in biocatalytic investigations. In Protein Engineering; Springer: Berlin, Germany, 2018; pp. 43–67. [Google Scholar]
- Mitra, S.; Dash, R. Structural dynamics and quantum mechanical aspects of shikonin derivatives as CREBBP bromodomain inhibitors. J. Mol. Graphics Modell. 2018, 83, 42–52. [Google Scholar] [CrossRef]
- Dash, R.; Das, R.; Junaid, M.; Akash, M.F.C.; Islam, A.; Hosen, S.Z. In silico-based vaccine design against Ebola virus glycoprotein. Adv.Appl. Bioinform. Chem. AABC 2017, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The amber lipid force field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef]
- Stewart, J.J. MOPAC: A semiempirical molecular orbital program. J. Comput. Aided Mol. Des. 1990, 4, 1–103. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Harrach, M.F.; Drossel, B. Structure and dynamics of TIP3P, TIP4P, and TIP5P water near smooth and atomistic walls of different hydroaffinity. J. Chem. Phys. 2014, 140, 174501. [Google Scholar] [CrossRef] [PubMed]
- Shaik, M.S.; Liem, S.Y.; Popelier, P.L. Properties of liquid water from a systematic refinement of a high-rank multipolar electrostatic potential. J. Chem. Phys. 2010, 132, 174504. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Florová, P.; Sklenovský, P.; Banáš, P.; Otyepka, M. Explicit water models affect the specific solvation and dynamics of unfolded peptides while the conformational behavior and flexibility of folded peptides remain intact. J. Chem. Theory Comput. 2010, 6, 3569–3579. [Google Scholar] [CrossRef]
- Fennell, C.J.; Kehoe, C.W.; Dill, K.A. Modeling aqueous solvation with semi-explicit assembly. Proc. Natl. Acad. Sci. USA 2011, 108, 3234–3239. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Dunbrack, R.L.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pK a prediction with hydrogen bonding network optimization. In Computational Drug Discovery and Design; Springer: Berlin, Germany, 2012; pp. 405–421. [Google Scholar]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Gr. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Modell. Simul. Mater. Sci. Eng. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Knowledge-based protein secondary structure assignment. Proteins Struct. Funct. Bioinform. 1995, 23, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. DSSP: Definition of secondary structure of proteins given a set of 3D coordinates. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Blatt, J.M.; Weisskopf, V.F. Theoretical Nuclear Physics; Courier Corporation: North Chelmsford, MA, USA, 1991. [Google Scholar]
- Lovering, A.L.; Lee, S.S.; Kim, Y.-W.; Withers, S.G.; Strynadka, N.C. Mechanistic and structural analysis of a family 31 a-glycosidase and its glycosyl-enzyme intermediate. J. Biol. Chem. 2004, 280, 2105–2115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, E.; Rajasekaran, R. Computational investigation of curcumin, a natural polyphenol that inhibits the destabilization and the aggregation of human SOD1 mutant (Ala4Val). RSC Adv. 2016, 6, 102744–102753. [Google Scholar] [CrossRef]
- Swanson, J.M.; Henchman, R.H.; McCammon, J.A. Revisiting free energy calculations: A theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys. J. 2004, 86, 67–74. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [Green Version]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins Struct. Funct. Bioinform. 1991, 11, 205–217. [Google Scholar] [CrossRef]
- Tripathi, S.; Srivastava, G.; Sharma, A. Molecular dynamics simulation and free energy landscape methods in probing L215H, L217R and L225M βI-tubulin mutations causing paclitaxel resistance in cancer cells. Biochem. Biophys. Res. Commun. 2016, 476, 273–279. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, Y.-Q.; Ai, W.-B.; Hu, Q.-T.; Zhang, Q.-J.; Wan, L.-Y.; Wang, X.-L.; Liu, C.-B.; Wu, J.-F. Hes1, an important gene for activation of hepatic stellate cells, is regulated by Notch1 and TGF-β/BMP signaling. World J. Gastroenterol. WJG 2015, 21, 878. [Google Scholar] [CrossRef]
- Shlens, J. A tutorial on principal component analysis. arXiv 2014, arXiv:1404.1100. [Google Scholar]
- Salmas, R.E.; Yurtsever, M.; Durdagi, S. Investigation of inhibition mechanism of chemokine receptor CCR5 by micro-second molecular dynamics simulations. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, O.F.; Grubmüller, H. Generalized correlation for biomolecular dynamics. Proteins Struct. Funct. Bioinform. 2006, 62, 1053–1061. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Leonis, G.; Steinbrecher, T.; Papadopoulos, M.G. A contribution to the drug resistance mechanism of Darunavir, Amprenavir, Indinavir, and Saquinavir complexes with HIV-1 protease due to flap mutation I50V: A systematic MM–PBSA and thermodynamic integration study. J. Chem. Inf. Model. 2013, 53, 2141–2153. [Google Scholar] [CrossRef]
- Chen, J.; Wang, X.; Zhu, T.; Zhang, Q.; Zhang, J.Z. A comparative insight into amprenavir resistance of mutations V32I, G48V, I50V, I54V, and I84V in HIV-1 protease based on thermodynamic integration and MM-PBSA methods. J. Chem. Inf. Model. 2015, 55, 1903–1913. [Google Scholar] [CrossRef]
- Musyoka, T.M.; Kanzi, A.M.; Lobb, K.A.; Bishop, Ö.T. Structure based docking and molecular dynamic studies of plasmodial cysteine proteases against a South African natural compound and its analogs. Sci. Rep. 2016, 6, 23690. [Google Scholar] [CrossRef] [Green Version]
- Moonrin, N.; Songtawee, N.; Rattanabunyong, S.; Chunsrivirot, S.; Mokmak, W.; Tongsima, S.; Choowongkomon, K. Understanding the molecular basis of EGFR kinase domain/MIG-6 peptide recognition complex using computational analyses. BMC Bioinform. 2015, 16, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | MM-PBSA $ ∆Gbinding | CASTp & | MM-PBSA # ∆Gbinding | |||||
|---|---|---|---|---|---|---|---|---|
| (kJ/mol) | Volume | SASA | ΔEvdW (kJ/mol) | ΔEelec (kJ/mol) | ΔGpol (kJ/mol) | ΔGnonpo (kJ/mol) | ΔGBinding (kJ/mol) | |
| Wild | 453.31 | 810.58 | − | − | − | − | − | |
| R213H | 596.88 | 1020.92 | − | − | − | − | − | |
| Wild-PNP | 110 ± 1.03 | 336.87 | 537.45 | −80.18 ± 0.27 | −8.23± 0.54 | 77.80 ± 0.36 | −89.84 ± 0.30 | −120.42 ± 0.68 |
| R213H-PNP | 60 ± 2.46 | 236.66 | 607.83 | −73.25 ± 0.27 | −24.06 ± 0.54 | 66.27 ± 0.56 | −83.99 ± 0.34 | −115.00 ± 0.62 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, R.; Ali, M.C.; Dash, N.; Azad, M.A.K.; Hosen, S.M.Z.; Hannan, M.A.; Moon, I.S. Structural and Dynamic Characterizations Highlight the Deleterious Role of SULT1A1 R213H Polymorphism in Substrate Binding. Int. J. Mol. Sci. 2019, 20, 6256. https://doi.org/10.3390/ijms20246256
Dash R, Ali MC, Dash N, Azad MAK, Hosen SMZ, Hannan MA, Moon IS. Structural and Dynamic Characterizations Highlight the Deleterious Role of SULT1A1 R213H Polymorphism in Substrate Binding. International Journal of Molecular Sciences. 2019; 20(24):6256. https://doi.org/10.3390/ijms20246256
Chicago/Turabian StyleDash, Raju, Md. Chayan Ali, Nayan Dash, Md. Abul Kalam Azad, S. M. Zahid Hosen, Md. Abdul Hannan, and Il Soo Moon. 2019. "Structural and Dynamic Characterizations Highlight the Deleterious Role of SULT1A1 R213H Polymorphism in Substrate Binding" International Journal of Molecular Sciences 20, no. 24: 6256. https://doi.org/10.3390/ijms20246256