Candidate Gene Selection for Cytoplasmic Male Sterility in Pepper (Capsicum annuum L.) through Whole Mitochondrial Genome Sequencing

Abstract
:1. Introduction
2. Results
2.1. Mitochondrial Genome Sequence of CMS Line 138A and Its Maintainer Line 138B
2.2. ORF Identification and Gene Annotation
2.3. Comparative Analysis of the Mitochondrial Genomes
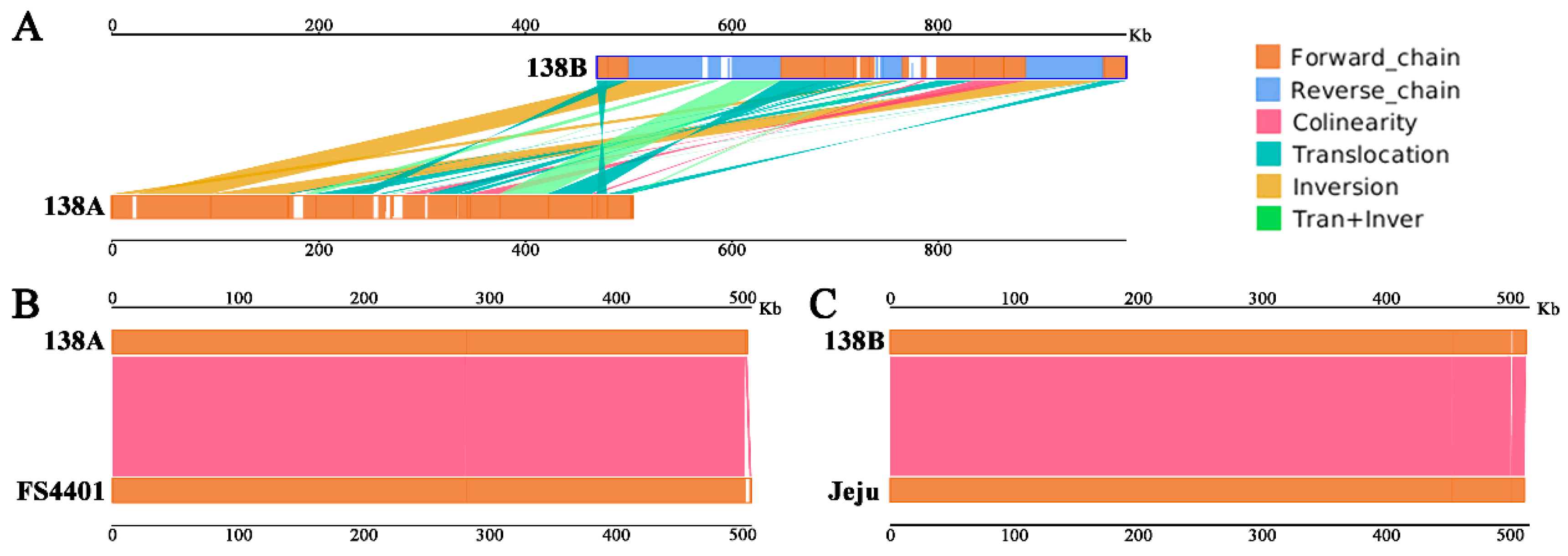
2.3.1. Syntenic Sequence Analysis of the Mitochondrial Genomes
2.3.2. Genome Structural Variation between 138A and 138B
2.3.3. SNP and InDel Detection
2.4. Selection of Candidate Genes Controlling the Cytoplasmic Male Sterility
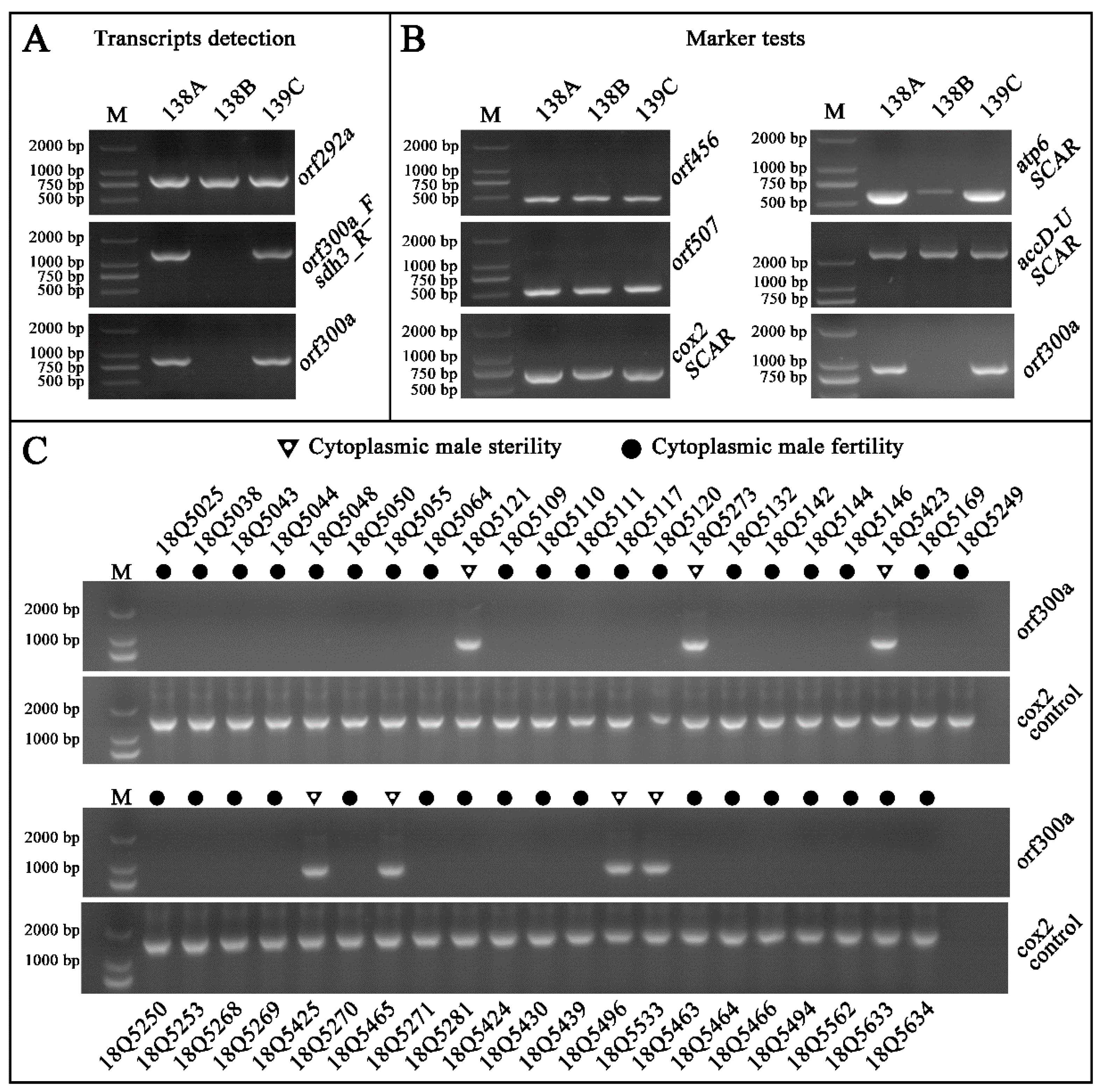
2.4.1. Identification of Novel ORFs in the Unique Regions of 138A
2.4.2. Analyses of Chimeric Structures, Co-Transcript Event, and Transmembrane Domain
2.5. CMS Marker Development and Testing
3. Discussion
3.1. 138A and FS4401 May Originate from a Common Female Ancestor, but Their CMS May Originate Separately
3.2. Candidate Gene Selection for the CMS Trait in 138A
4. Materials and Methods
4.1. Plant Materials
4.2. Mitochondrial DNA Extraction
4.3. Sequencing and Assembling of the Mitochondrial Genome
4.4. Genome Component Analysis
4.4.1. Gene Annotation and Identification of ORFs
4.4.2. Sequence Comparison between 138A and 138B
4.4.3. RT–PCR, Real-time PCR, and Sequencing of PCR Products
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tester, M.; Langridge, P. Breeding technologies to increase crop production in a changing world. Science 2010, 327, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, D.; Zhu, Y. Characterization and use of male sterility in hybrid rice breeding. J. Integr. Plant Biol. 2007, 49, 791–804. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Y.G. Male Sterility and Fertility Restoration in Crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Wise, R.P. The molecular basis of cytoplasmic male sterility and fertility restoration. Trends Plant Sci. 1998, 3, 175–180. [Google Scholar] [CrossRef]
- Hanson, M.R.; Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 2004, 16 (Suppl. 1), S154–S169. [Google Scholar] [CrossRef]
- Knoop, V. The mitochondrial DNA of land plants: Peculiarities in phylogenetic perspective. Curr. Genet. 2004, 46, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, I.E. Mitochondria; Wiley-Liss: New York, NY, USA, 1999. [Google Scholar]
- Palmer, J.D.; Herbon, L.A. Unicircular structure of the Brassica hirta mitochondrial genome. Curr. Genet. 1987, 11, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Jo, Y.D.; Choi, Y.; Kim, D.H.; Kim, B.D.; Kang, B.C. Extensive structural variations between mitochondrial genomes of CMS and normal peppers (Capsicum annuum L.) revealed by complete nucleotide sequencing. BMC Genomics. 2014, 15, 561. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid evolution of enormous, multichromosomal genomes in flowering plant mitochondria with exceptionally high mutation rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef]
- Budar, F.; Touzet, P.; De Paepe, R. The nucleo-mitochondrial conflict in cytoplasmic male sterilities revised. Genetica 2003, 117, 3–16. [Google Scholar] [CrossRef]
- Kubo, T.; Newton, K.J. Angiosperm mitochondrial genomes and mutations. Mitochondrion 2008, 8, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Small, I.; Suffolk, R.; Leaver, C.J. Evolution of plant mitochondrial genomes via substoichiometric intermediates. Cell 1989, 58, 69–76. [Google Scholar] [CrossRef]
- Albert, B.; Godelle, B.; Gouyon, P.H. Evolution of the plant mitochondrial genome: Dynamics of duplication and deletion of sequences. J. Mol. Evol. 1998, 46, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Woloszynska, M.; Trojanowski, D. Counting mtDNA molecules in Phaseolus vulgaaris: Sublimons are constantly produced by recombination via short repeats and undergo rigorous selection during substoichiometric shifting. Plant Mol. Biol. 2009, 70, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Mayr, E. Joseph Gottlieb Kolreuter’s contributions to biology. Osiris 1986, 2, 76–135. [Google Scholar] [CrossRef]
- Kaul, M.L. Male Sterility in Higher Plants; Springer: New York, NY, USA, 2012. [Google Scholar]
- Wang, Z.; Zou, Y.; Li, X.; Zhang, Q.; Chen, L.; Wu, H.; Su, D.; Chen, Y.L.; Guo, J.X.; Luo, D.; et al. Cytoplasmic male sterility of rice with boro II cytoplasm is caused by a cytotoxic peptide and is restored by two related PPR motif genes via distinct modes of mRNA silencing. Plant Cell 2006, 18, 676–687. [Google Scholar] [CrossRef]
- Park, J.Y.; Lee, Y.P.; Lee, J.; Choi, B.S.; Kim, S.; Yang, T.J. Complete mitochondrial genome sequence and identification of a candidate gene responsible for cytoplasmic male sterility in radish (Raphanus sativus L.) containing DCGMS cytoplasm. Theor. Appl. Genet. 2013, 126, 1763–1774. [Google Scholar] [CrossRef]
- Okazaki, M.; Kazama, T.; Murata, H.; Motomura, K.; Toriyama, K. Whole mitochondrial genome sequencing and transcriptional analysis to uncover an RT102-type cytoplasmic male sterility-associated candidate gene derived from Oryza rufipogon. Plant Cell Physiol. 2013, 54, 1560–1568. [Google Scholar] [CrossRef]
- Cui, X.; Wise, R.P.; Schnable, P.S. The rf2 nuclear restorer gene of male-sterile T-cytoplasm maize. Science 1996, 272, 1334–1336. [Google Scholar] [CrossRef]
- Liu, F.; Cui, X.; Horner, H.T.; Weiner, H.; Schnable, P.S. Mitochondrial aldehyde dehydrogenase activity is required for male fertility in maize. Plant Cell 2001, 13, 1063–1078. [Google Scholar] [CrossRef]
- Fujii, S.; Komatsu, S.; Toriyama, K. Retrograde regulation of nuclear gene expression in CW-CMS of rice. Plant Mol. Biol. 2007, 63, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Toriyama, K. Suppressed expression of retrograde-regulated male sterility restores pollen fertility in cytoplasmic male sterile rice plants. Proc. Natl. Acad. Sci. USA 2009, 106, 9513–9518. [Google Scholar] [CrossRef] [PubMed]
- Bentolila, S.; Alfonso, A.A.; Hanson, M.R. A pentatricopeptide repeat-containing gene restores fertility to cytoplasmic male-sterile plants. Proc. Natl. Acad. Sci. USA 2002, 99, 10887–10892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, Y.D.; Ha, Y.; Lee, J.H.; Park, M.; Bergsma, A.C.; Choi, H.I.; Goritschnig, S.; Kloosterman, B.; van Dijk, P.J.; Choi, D.; et al. Fine mapping of restorer-of-fertility in pepper (Capsicum annuum L.) identified a candidate gene encoding a pentatricopeptide repeat (PPR)-containing protein. Theor. Appl. Genet. 2016, 129, 2003–2017. [Google Scholar] [CrossRef]
- Delannoy, E.; Stanley, W.A.; Bond, C.S.; Small, I.D. Pentatricopeptide repeat (PPR) proteins as sequence-specificity factors in post-transcriptional processes in organelles. Biochem. Soc. Trans. 2007, 35, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Linneweber, C.; Small, I. Pentatricopeptide repeat proteins: A socket set for organelle gene expression. Trends Plant Sci. 2008, 13, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kang, J.G.; Kim, S.; Kim, B.D. Identification of cox II and atp6 region as associated to CMS in Capsicum annuum by using RFLP and long and accurate PCR. Hortic. Environ. Biotech. 2001, 42, 121–127. [Google Scholar]
- Kim, D.H.; Kim, B.D. Development of SCAR markers for early identification of cytoplasmic male sterility genotype in chili pepper (Capsicum annuum L.). Mol. Cell 2005, 20, 416–422. [Google Scholar]
- Kim, D.H.; Kim, B.D. The organization of mitochondrial atp6 gene region in male fertile and CMS lines of pepper (Capsicum annuum L.). Curr. Genet. 2006, 49, 59–67. [Google Scholar] [CrossRef]
- Kim, D.H.; Kang, J.G.; Kim, B.D. Isolation and characterization of the cytoplasmic male sterility-associated orf456 gene of chili pepper (Capsicum annuum L.). Plant Mol. Biol. 2007, 63, 519–532. [Google Scholar] [CrossRef]
- Gulyas, G.; Shin, Y.; Kim, H.; Lee, J.S.; Hirata, Y. Altered Transcript Reveals an Orf507 Sterility-Related Gene in Chili Pepper (Capsicum annuum L.). Plant Mol. Biol. Rep. 2010, 28, 605–612. [Google Scholar] [CrossRef]
- Jo, Y.K.; Jeong, H.J.; Kang, B.C. Development of a CMS-specific marker based on chloroplast-derived mitochondrial sequence in pepper. Plant Biotechnol. Rep. 2009, 3, 309–315. [Google Scholar] [CrossRef]
- Shen, H.L.; Jiang, J.Z.; Wang, Z.; Geng, S.S. Studies on the breeding and inheritance of male sterile lines of pepper (Capsicum annuum L.). J. Beijing Agric. Univ. 1994, 20, 25–30. [Google Scholar]
- Lee, J.H.; An, J.T.; Han, K.; Choi, S.; Siddique, M.I.; Kang, B.C. Genetic mapping of resistance sources against ChiVMV (Chili veinal mottle virus) in hot pepper. Gene Genome New Technol. Plant Breed. 2016, 1, 220. [Google Scholar]
- Wang, R.; Cai, X.H.; Fan, Y.J.; Hu, S.G.; Zhou, W. Advances in Solanaceae Mitochondrial Genomics. Genom. App. Biol. 2018, 1–10. [Google Scholar]
- Heazlewood, J.L.; Whelan, J.; Millar, A.H. The products of the mitochondrial orf25 and orfB genes are F0 components in the plant F1F0 ATP synthase. FEBS Lett. 2003, 540, 201–205. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, T.; Zhao, T.; Huang, H. Aberrant mitochondrial gene orf25 may cause tobacco male sterility. Acta Agric. Univ. Jiangxiensi 2009, 31, 54–62. [Google Scholar]
- Zhou, W.; Huang, H.; Zhou, B.N.; Liu, Q.Y. The bioinformatics analysis of orf25 gene related to tobacco cytoplasmic male sterility. Chin. Agric. Sci. Bull. 2011, 5, 312–316. [Google Scholar]
- Bonhomme, S.; Budar, F.; Lancelin, D.; Small, I.; Defrance, M.; Pelletier, G. Sequence and transcript analysis of the Nco2.5 Ogura-specific fragment correlated with cytoplasmic male sterility in Brassica cybrids. Mol. Gen. Genet. 1992, 235, 340–348. [Google Scholar] [CrossRef]
- Tanaka, Y.; Tsuda, M.; Yasumoto, K.; Yamagishi, H.; Terachi, T. A complete mitochondrial genome sequence of Ogura-type male-sterile cytoplasm and its comparative analysis with that of normal cytoplasm in radish (Raphanus sativus L.). BMC Genom. 2012, 13, 352. [Google Scholar] [CrossRef]
- Tang, H.V.; Pring, D.R.; Shaw, L.C.; Salazar, R.A.; Muza, F.R.; Yan, B.; Schertz, K.F. Transcript processing internal to a mitochondrial open reading frame is correlated with fertility restoration in male-sterile sorghum. Plant J. 1996, 10, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, R.H.; Horn, R.; Lössl, A.; Zetsche, K. Cytoplasmic male sterility in sunflower is correlated with the co-transcription of a new open reading frame with the atpA gene. Mol. Gen. Genet. 1991, 227, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Borgström, E.; Lundin, S.; Lundeberg, J. Large scale library generation for high throughput sequencing. PLoS One 2011, 6, e19119. [Google Scholar] [CrossRef]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. HYBRIDSPADES: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome. Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32 (Suppl. 1), D277–D280. [Google Scholar] [CrossRef]
- Kanehisa, M. A database for post-genome analysis. Trends Genet. 1997, 13, 375. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34 (Suppl. 1), D354–D357. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Magrane, M. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, 9. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. Organellar Genome DRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features of the ORF | ORF ID |
|---|---|
| SNP/InDel | orf229a, orf138a, orf337a, orf675a, orf249a, orf104c, orfB, orf25 |
| On the unique region | orf108g, orf132a, orf314a, orf262a, orf165a, orf338a, orf244a, orf100b, orf300a, orf119a, orf100a |
| Chimeric structure/ co-transcription | orf292a, orf300a |
| Transmembrane domain | orf300a, orf314a, orf157a, orf115b, orf262a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, P.; Lu, Q.; Ai, Y.; Wang, Y.; Li, T.; Wu, L.; Liu, J.; Cheng, Q.; Sun, L.; Shen, H. Candidate Gene Selection for Cytoplasmic Male Sterility in Pepper (Capsicum annuum L.) through Whole Mitochondrial Genome Sequencing. Int. J. Mol. Sci. 2019, 20, 578. https://doi.org/10.3390/ijms20030578
Wang P, Lu Q, Ai Y, Wang Y, Li T, Wu L, Liu J, Cheng Q, Sun L, Shen H. Candidate Gene Selection for Cytoplasmic Male Sterility in Pepper (Capsicum annuum L.) through Whole Mitochondrial Genome Sequencing. International Journal of Molecular Sciences. 2019; 20(3):578. https://doi.org/10.3390/ijms20030578
Chicago/Turabian StyleWang, Peng, Qiaohua Lu, Yixin Ai, Yihao Wang, Tiantian Li, Lang Wu, Jinqiu Liu, Qing Cheng, Liang Sun, and Huolin Shen. 2019. "Candidate Gene Selection for Cytoplasmic Male Sterility in Pepper (Capsicum annuum L.) through Whole Mitochondrial Genome Sequencing" International Journal of Molecular Sciences 20, no. 3: 578. https://doi.org/10.3390/ijms20030578