Epigenome-Wide Association Study (EWAS) of Blood Lipids in Healthy Population from STANISLAS Family Study (SFS)

 , ,
, ,
Abstract
:
1. Introduction
2. Results
2.1. Associations between Genome-Wide DNA Methylation and Lipid Levels in Whole Blood
2.2. Bioinformatics Analysis
2.3. Associations between Genetic Variants and Methylation Sites
2.4. Replication of the Identified CpGs Associations with Lipids Levels in Adipose Tissue
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Data Collection
4.3. Biological Measurements
4.4. DNA Methylation Assessment
4.4.1. Discovery Cohort
4.4.2. Replication Cohort
4.5. Genotyping and Selection of Single-Nucleotide Polymorphisms (SNPs)
4.6. Statistical Analysis
4.7. In Silico Analysis
4.8. Data Access
Author Contributions
Funding
Conflicts of Interest
References
- Vanuzzo, D.; Pilotto, L.; Mirolo, R.; Pirelli, S. Cardiovascular Risk and Cardiometabolic Risk: An Epidemiological Evaluation. G. Ital. Cardiol. (Rome) 2008, 9 (Suppl. 1), 6s–17s. [Google Scholar]
- Nayak, S.B.; Mohammed, S.B.; Nayak, A.S. Controlling Lipids Aids in the Prevention of Type 2 Diabetes, Hypertension, and Cardiovascular Diseases. Int. J. Prev. Med. 2017, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.J.; Hulley, S.B.; Browner, W.S.; Kuller, L.H.; Wentworth, D. Serum Cholesterol, Blood Pressure, and Mortality: Implications from a Cohort of 361,662 Men. Lancet 1986, 2, 933–936. [Google Scholar] [CrossRef]
- Arsenault, B.J.; Boekholdt, S.M.; Kastelein, J.J. Lipid Parameters for Measuring Risk of Cardiovascular Disease. Nat. Rev. Cardiol. 2011, 8, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Hokanson, J.E.; Austin, M.A. Plasma Triglyceride Level Is a Risk Factor for Cardiovascular Disease Independent of High-Density Lipoprotein Cholesterol Level: A Meta-Analysis of Population-Based Prospective Studies. J. Cardiovasc. Risk 1996, 3, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Pirani, N.; Khiavi, F.F. Population Attributable Fraction for Cardiovascular Diseases Risk Factors in Selected Countries: A Comparative Study. Mater. Sociomed. 2017, 29, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Faul, F.; Erdfelder, E.; Buchner, A.; Lang, A.G. Statistical Power Analyses Using G*Power 3.1: Tests for Correlation and Regression Analyses. Behav. Res. Methods 2009, 41, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, Clinical and Population Relevance of 95 Loci for Blood Lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and Refinement of Loci Associated with Lipid Levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Strong, A.; Frank-Kamenetsky, M.; Lee, N.E.; Ahfeldt, T.; Sachs, K.V.; Li, X.; Li, H.; Kuperwasser, N.; Ruda, V.M.; et al. From Noncoding Variant to Phenotype Via Sort1 at the 1p13 Cholesterol Locus. Nature 2010, 466, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-Wide Association Studies for Common Human Diseases. Nat. Rev. Genet. 2011, 12, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic Modifications: Basic Mechanisms and Role in Cardiovascular Disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA Methylation: Islands, Start Sites, Gene Bodies and Beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Hedman, A.K.; Mendelson, M.M.; Marioni, R.E.; Gustafsson, S.; Joehanes, R.; Irvin, M.R.; Zhi, D.; Sandling, J.K.; Yao, C.; Liu, C.; et al. Epigenetic Patterns in Blood Associated with Lipid Traits Predict Incident Coronary Heart Disease Events and Are Enriched for Results from Genome-Wide Association Studies. Circ. Cardiovasc. Genet. 2017, 10, e001487. [Google Scholar] [CrossRef] [PubMed]
- Sayols-Baixeras, S.; Irvin, M.R.; Arnett, D.K.; Elosua, R.; Aslibekyan, S.W. Epigenetics of Lipid Phenotypes. Curr. Cardiovasc. Risk Rep. 2016, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.V.; Voortman, T.; Dhana, K.; Troup, J.; Bramer, W.M.; Troup, J.; Chowdhury, R.; Dehghan, A.; Muka, T.; Franco, O.H. The Role of DNA Methylation in Dyslipidaemia: A Systematic Review. Prog. Lipid Res. 2016, 64, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, L.; Wahl, S.; Pilling, L.C.; Reischl, E.; Sandling, J.K.; Kunze, S.; Holdt, L.M.; Kretschmer, A.; Schramm, K.; Adamski, J.; et al. DNA Methylation of Lipid-Related Genes Affects Blood Lipid Levels. Circ. Cardiovasc. Genet. 2015, 8, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, K.V.E.; Dhana, K.; de Vries, P.S.; Voortman, T.; van Meurs, J.B.J.; Uitterlinden, A.G.; Hofman, A.; Hu, F.B.; Franco, O.H.; Dehghan, A. Epigenome-Wide Association Study (Ewas) on Lipids: The Rotterdam Study. Clin. Epigenet. 2017, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, K.F.; van Iterson, M.; Slieker, R.C.; Moed, M.H.; Bonder, M.J.; van Galen, M.; Mei, H.; Zhernakova, D.V.; van den Berg, L.H.; Deelen, J.; et al. Blood Lipids Influence DNA Methylation in Circulating Cells. Genome Biol. 2016, 17, 138. [Google Scholar] [CrossRef] [PubMed]
- Burwinkel, B.; Scott, J.W.; Buhrer, C.; van Landeghem, F.K.; Cox, G.F.; Wilson, C.J.; Hardie, D.G.; Kilimann, M.W. Fatal Congenital Heart Glycogenosis Caused by a Recurrent Activating R531q Mutation in the Gamma 2-Subunit of Amp-Activated Protein Kinase (Prkag2), Not by Phosphorylase Kinase Deficiency. Am. J. Hum. Genet. 2005, 76, 1034–1049. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Carling, D. The Amp-Activated Protein Kinase--Fuel Gauge of the Mammalian Cell? Eur. J. Biochem. 1997, 246, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Blair, E.; Redwood, C.; Ashrafian, H.; Oliveira, M.; Broxholme, J.; Kerr, B.; Salmon, A.; Ostman-Smith, I.; Watkins, H. Mutations in the Gamma(2) Subunit of Amp-Activated Protein Kinase Cause Familial Hypertrophic Cardiomyopathy: Evidence for the Central Role of Energy Compromise in Disease Pathogenesis. Hum. Mol. Genet. 2001, 10, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Benson, D.W.; Perez-Atayde, A.R.; McKenna, W.J.; Sparks, E.A.; Kanter, R.J.; McGarry, K.; Seidman, J.G.; Seidman, C.E. Constitutively Active Amp Kinase Mutations Cause Glycogen Storage Disease Mimicking Hypertrophic Cardiomyopathy. J. Clin. Investig. 2002, 109, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Arad, M.; Maron, B.J.; Gorham, J.M.; Johnson, W.H., Jr.; Saul, J.P.; Perez-Atayde, A.R.; Spirito, P.; Wright, G.B.; Kanter, R.J.; Seidman, C.E.; et al. Glycogen Storage Diseases Presenting as Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2005, 352, 362–372. [Google Scholar] [CrossRef] [PubMed]
- GmbH, Genomatix. Genome Annotation and Regulation Analysis Tool. Available online: https://www.genomatix.de/ (accessed on 21 February 2019).
- De Ferrari, G.V.; Papassotiropoulos, A.; Biechele, T.; De-Vrieze, F.W.; Avila, M.E.; Major, M.B.; Myers, A.; Saez, K.; Henriquez, J.P.; Zhao, A.; et al. Common Genetic Variation within the Low-Density Lipoprotein Receptor-Related Protein 6 and Late-Onset Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2007, 104, 9434–9439. [Google Scholar] [CrossRef] [PubMed]
- Mani, A.; Radhakrishnan, J.; Wang, H.; Mani, A.; Mani, M.A.; Nelson-Williams, C.; Carew, K.S.; Mane, S.; Najmabadi, H.; Wu, D.; et al. Lrp6 Mutation in a Family with Early Coronary Disease and Metabolic Risk Factors. Science 2007, 315, 1278–1282. [Google Scholar] [CrossRef] [PubMed]
- Sarzani, R.; Salvi, F.; Bordicchia, M.; Guerra, F.; Battistoni, I.; Pagliariccio, G.; Carbonari, L.; Dessi-Fulgheri, P.; Rappelli, A. Carotid Artery Atherosclerosis in Hypertensive Patients with a Functional Ldl Receptor-Related Protein 6 Gene Variant. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Visvikis-Siest, S.; Siest, G. The Stanislas Cohort: A 10-Year Follow-up of Supposed Healthy Families. Gene-Environment Interactions, Reference Values and Evaluation of Biomarkers in Prevention of Cardiovascular Diseases. Clin. Chem. Lab. Med. 2008, 46, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Siest, G.; Visvikis, S.; Herbeth, B.; Gueguen, R.; Vincent-Viry, M.; Sass, C.; Beaud, B.; Lecomte, E.; Steinmetz, J.; Locuty, J.; et al. Objectives, Design and Recruitment of a Familial and Longitudinal Cohort for Studying Gene-Environment Interactions in the Field of Cardiovascular Risk: The Stanislas Cohort. Clin. Chem. Lab. Med. 1998, 36, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A Simple Salting out Procedure for Extracting DNA from Human Nucleated Cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [PubMed]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the Concentration of Low-Density Lipoprotein Cholesterol in Plasma, without Use of the Preparative Ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [PubMed]
- Noma, A.; Hata, Y.; Goto, Y. Quantitation of Serum Apolipoprotein a-I, a-Ii, B, C-Ii, C-Iii and E in Healthy Japanese by Turbidimetric Immunoassay: Reference Values, and Age- and Sex-Related Differences. Clin. Chim. Acta 1991, 199, 147–157. [Google Scholar] [CrossRef]
- Bibikova, M.; Barnes, B.; Tsan, C.; Ho, V.; Klotzle, B.; Le, J.M.; Delano, D.; Zhang, L.; Schroth, G.P.; Gunderson, K.L.; et al. High Density DNA Methylation Array with Single Cpg Site Resolution. Genomics 2011, 98, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of Cross-Reactive Probes and Polymorphic Cpgs in the Illumina Infinium Humanmethylation450 Microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, J.; Gordon, L.; Oshlack, A. Swan: Subset-Quantile within Array Normalization for Illumina Infinium Humanmethylation450 Beadchips. Genome Biol. 2012, 13, R44. [Google Scholar] [CrossRef] [PubMed]
- Grundberg, E.; Meduri, E.; Sandling, J.K.; Hedman, A.K.; Keildson, S.; Buil, A.; Busche, S.; Yuan, W.; Nisbet, J.; Sekowska, M.; et al. Global Analysis of DNA Methylation Variation in Adipose Tissue from Twins Reveals Links to Disease-Associated Variants in Distal Regulatory Elements. Am. J. Hum. Genet. 2013, 93, 876–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E. The New Nhgri-Ebi Catalog of Published Genome-Wide Association Studies (Gwas Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Barfield, R.T.; Kilaru, V.; Smith, A.K.; Conneely, K.N. Cpgassoc: An R Function for Analysis of DNA Methylation Microarray Data. Bioinformatics 2012, 28, 1280–1281. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, A.; Brockhoff, P.; Christensen, R.H.B. Lmertest: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017, 2. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at Ucsc. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
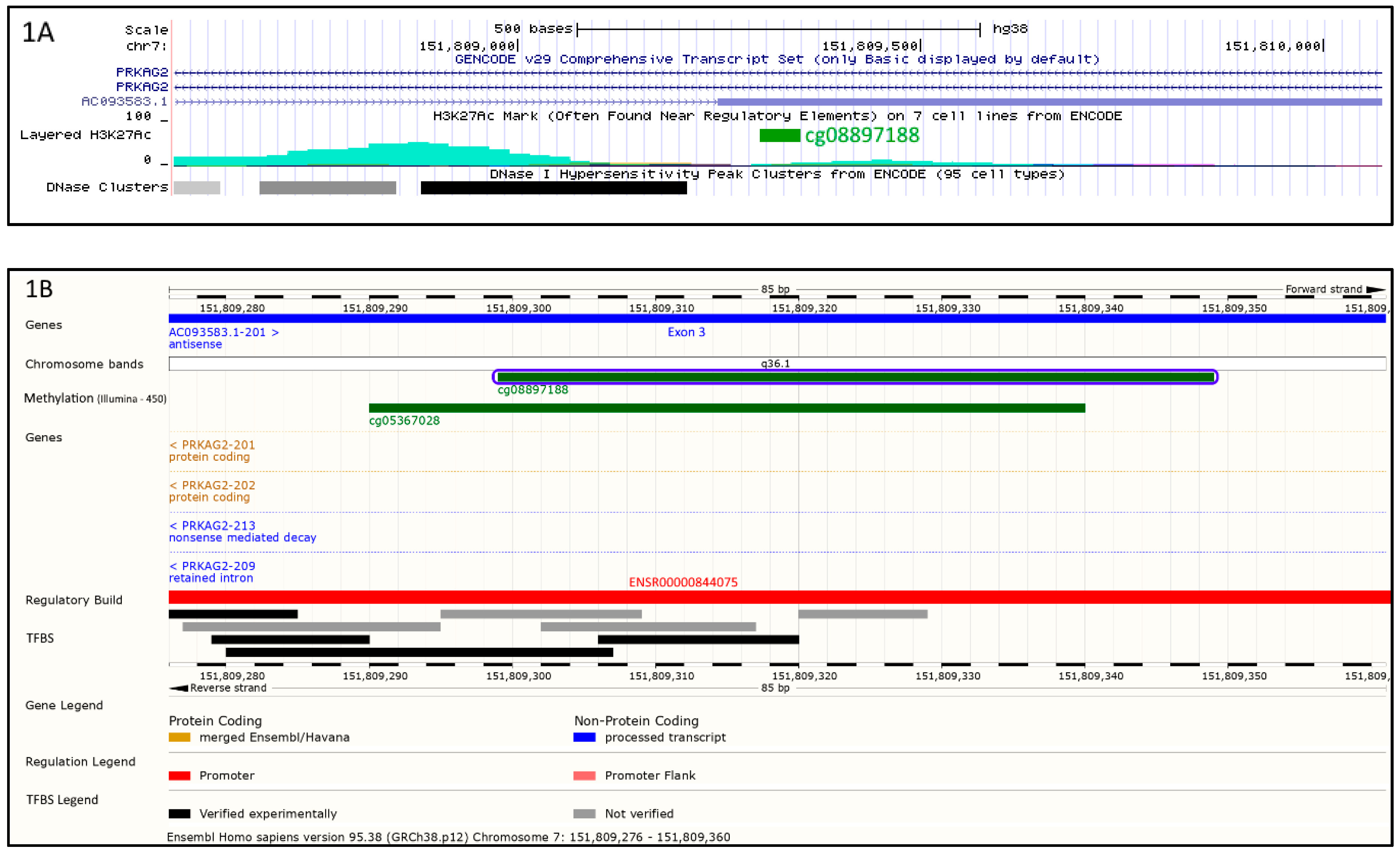

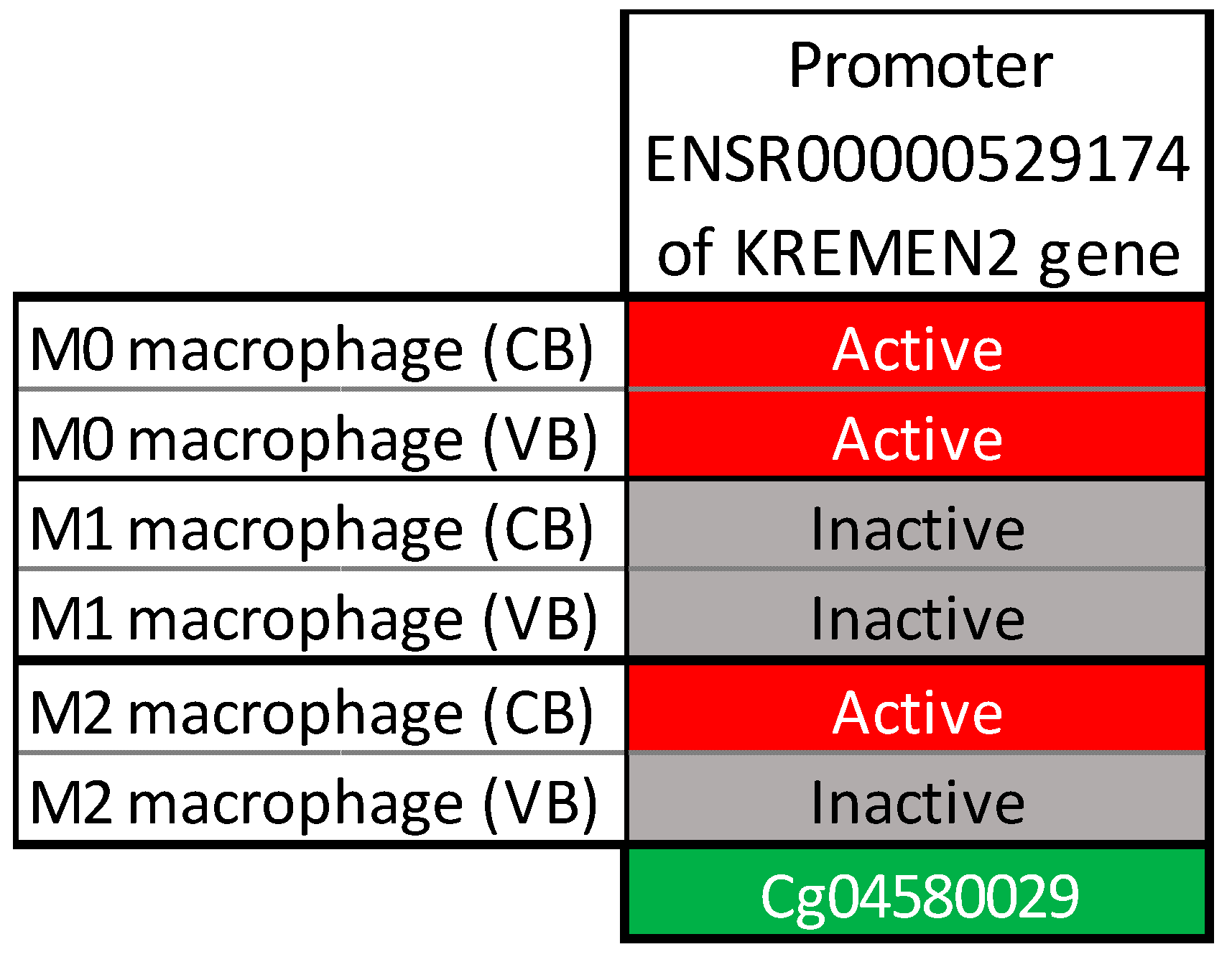
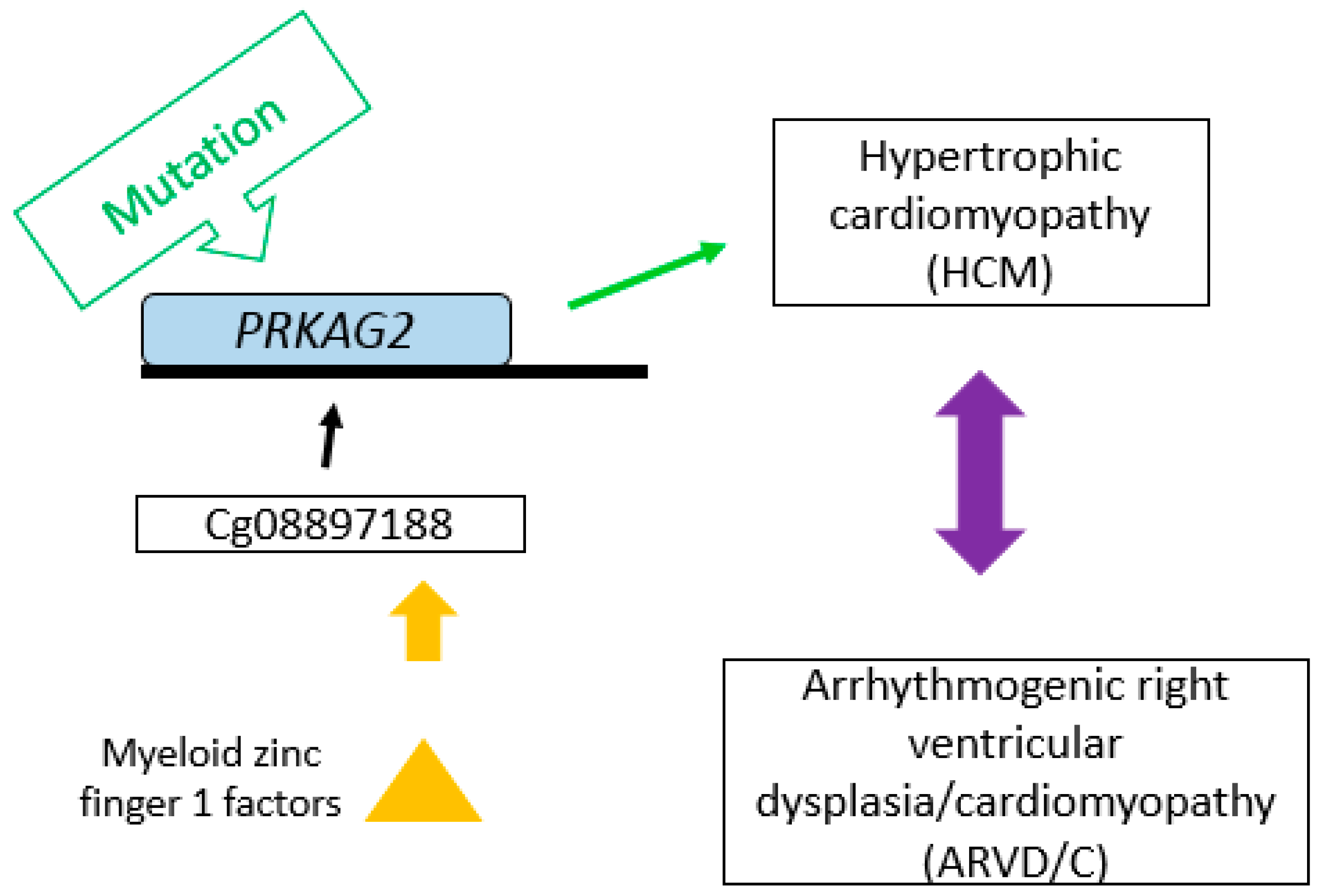

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CpG | Gene Name | Chromosome | Effect Size | SE | Unadjusted p-Value | FDR |
|---|---|---|---|---|---|---|
| cg08897188 | PRKAG2 | 7q36.1 | −2.80 | 0.47 | 1.39 × 10−8 | 0.049 |
| cg04580029 | KREMEN2 | 16p13.3 | 3.09 | 0.51 | 5.75 × 10−9 | 0.049 |
| CpG | Gene Name | Effect Size | SE | p-Value |
|---|---|---|---|---|
| cg08897188 | PRKAG2 | −0.0008 | 0.0016 | 0.6265 |
| cg04580029 | KREMEN2 | 0.0084 | 0.0036 | 0.0196 |
| Population Characteristics | Total | Adults (116) | Children (95) | Male (105) | Female (106) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | |
| Age (years) | 28.17 | 14.83 | 40.48 | 7.53 | 13.15 | 2.58 | 27.09 | 15.14 | 29.24 | 14.37 |
| Body Mass Index (kg/m2) | 21.52 | 4.00 | 24.06 | 3.22 | 18.43 | 2.31 | 21.77 | 4.17 | 21.27 | 3.79 |
| Cholesterol (mmol/L) | 5.24 | 1.00 | 5.70 | 0.96 | 4.68 | 0.74 | 5.12 | 1.04 | 5.37 | 0.95 |
| Triglycerides (mmol/L) | 0.86 | 0.49 | 0.98 | 0.56 | 0.72 | 0.32 | 0.91 | 0.55 | 0.82 | 0.41 |
| High density lipoprotein (mmol/L) | 1.44 | 0.41 | 1.45 | 0.42 | 1.43 | 0.39 | 1.34 | 0.38 | 1.54 | 0.42 |
| Low density lipoprotein (mmol/L) | 3.63 | 0.97 | 4.06 | 0.96 | 3.11 | 0.68 | 3.59 | 1.02 | 3.67 | 0.91 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, T.; Gorenjak, V.; G. Stathopoulou, M.; Dadé, S.; Marouli, E.; Masson, C.; Murray, H.; Lamont, J.; Fitzgerald, P.; Deloukas, P.; et al. Epigenome-Wide Association Study (EWAS) of Blood Lipids in Healthy Population from STANISLAS Family Study (SFS). Int. J. Mol. Sci. 2019, 20, 1014. https://doi.org/10.3390/ijms20051014
Xie T, Gorenjak V, G. Stathopoulou M, Dadé S, Marouli E, Masson C, Murray H, Lamont J, Fitzgerald P, Deloukas P, et al. Epigenome-Wide Association Study (EWAS) of Blood Lipids in Healthy Population from STANISLAS Family Study (SFS). International Journal of Molecular Sciences. 2019; 20(5):1014. https://doi.org/10.3390/ijms20051014
Chicago/Turabian StyleXie, Ting, Vesna Gorenjak, Maria G. Stathopoulou, Sébastien Dadé, Eirini Marouli, Christine Masson, Helena Murray, John Lamont, Peter Fitzgerald, Panagiotis Deloukas, and et al. 2019. "Epigenome-Wide Association Study (EWAS) of Blood Lipids in Healthy Population from STANISLAS Family Study (SFS)" International Journal of Molecular Sciences 20, no. 5: 1014. https://doi.org/10.3390/ijms20051014