Reaction Mechanisms and Kinetics of the Hydrogen Abstraction Reactions of C4–C6 Alkenes with Hydroxyl Radical: A Theoretical Exploration


Abstract
:1. Introduction
2. Results and Discussion
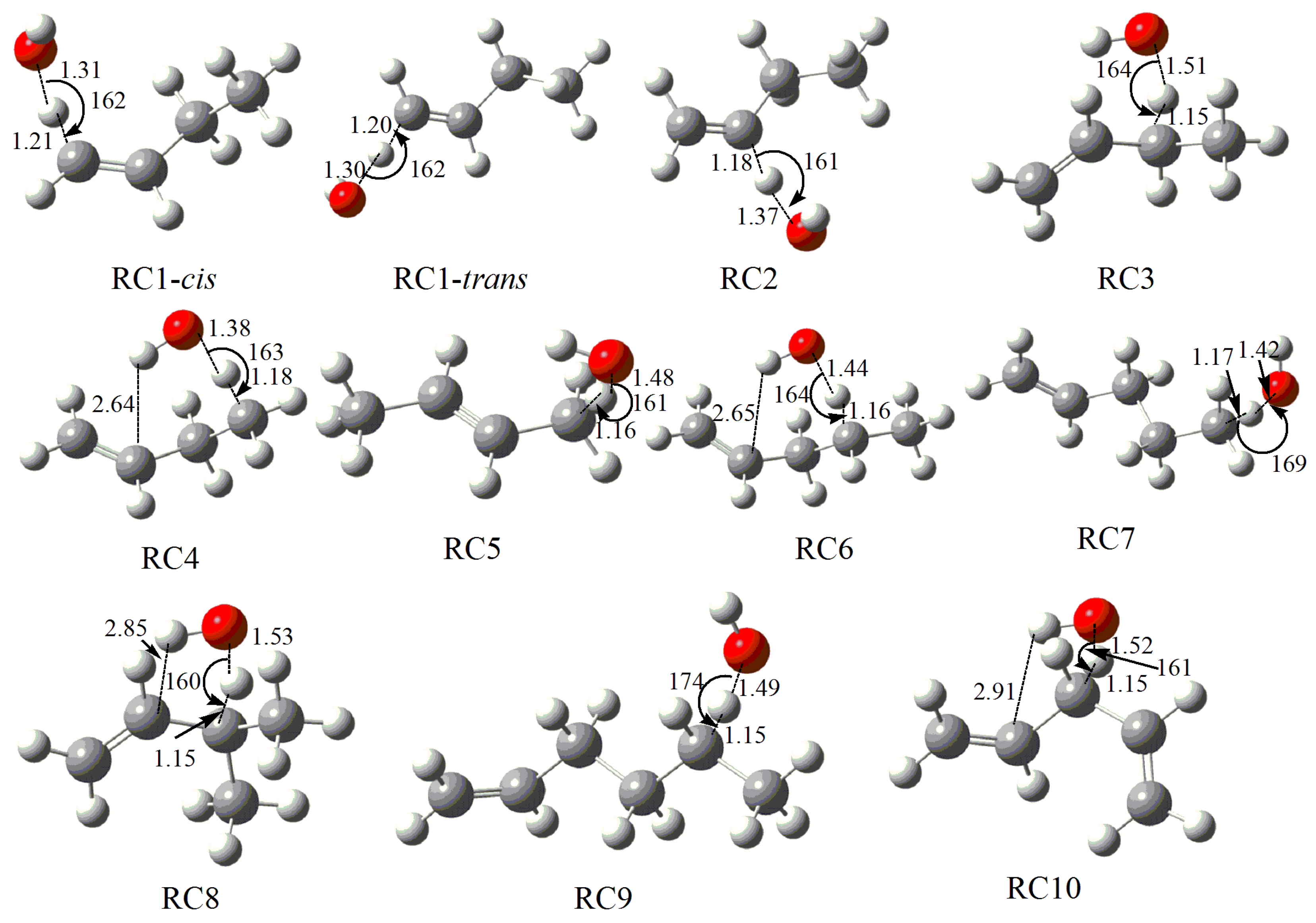
2.1. Geometry Analysis
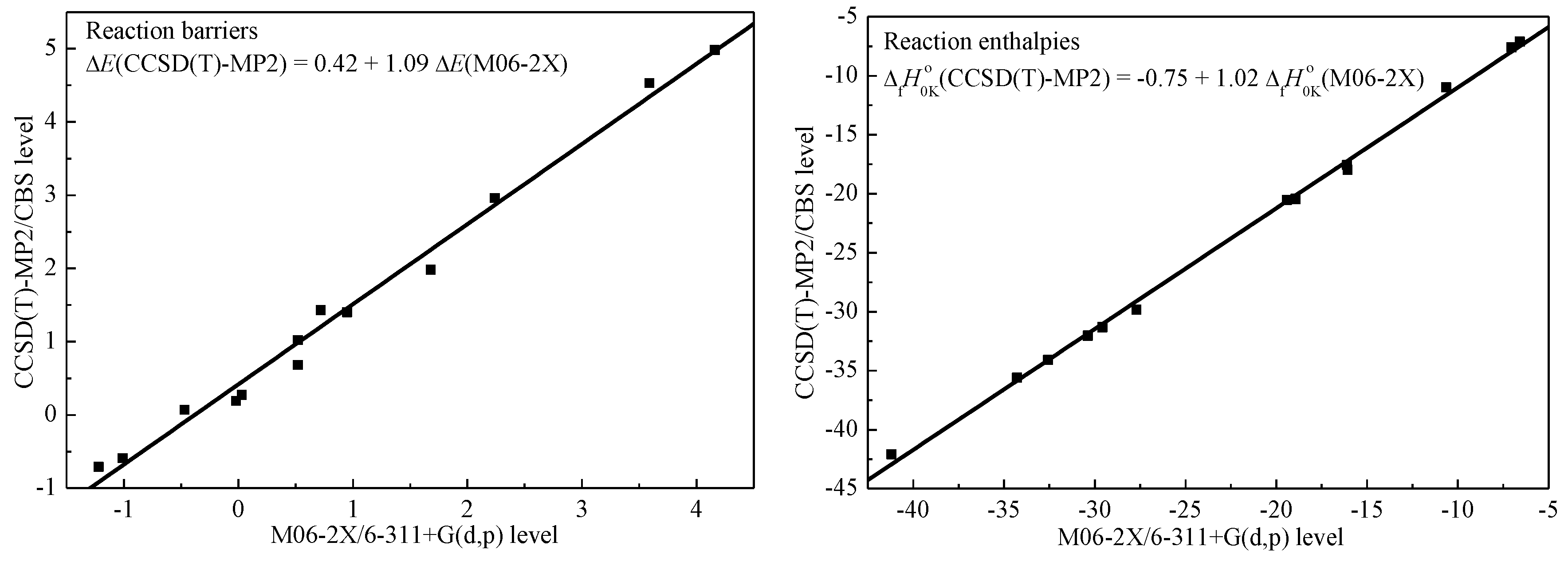
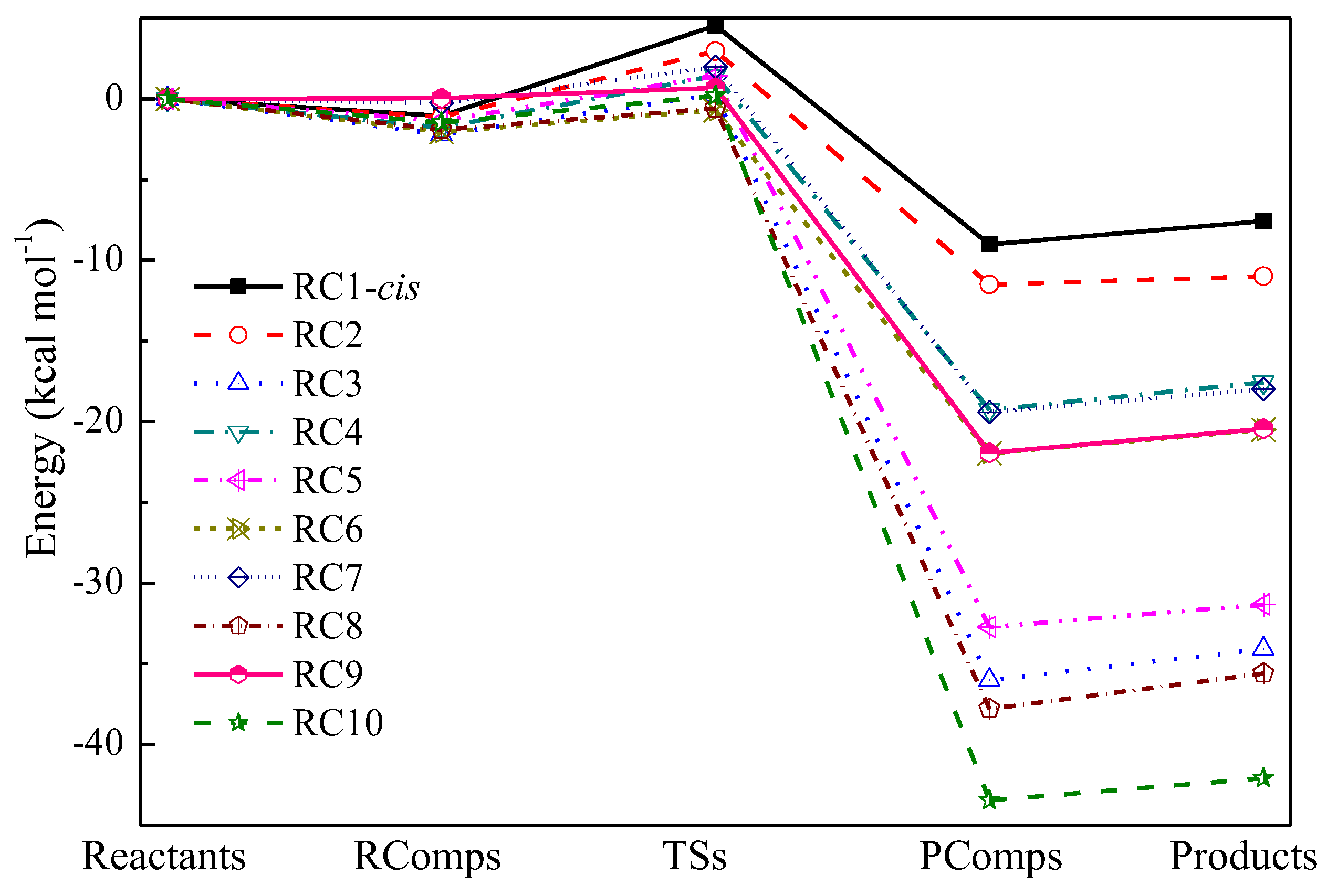
2.2. Reaction Barriers and Enthalpies
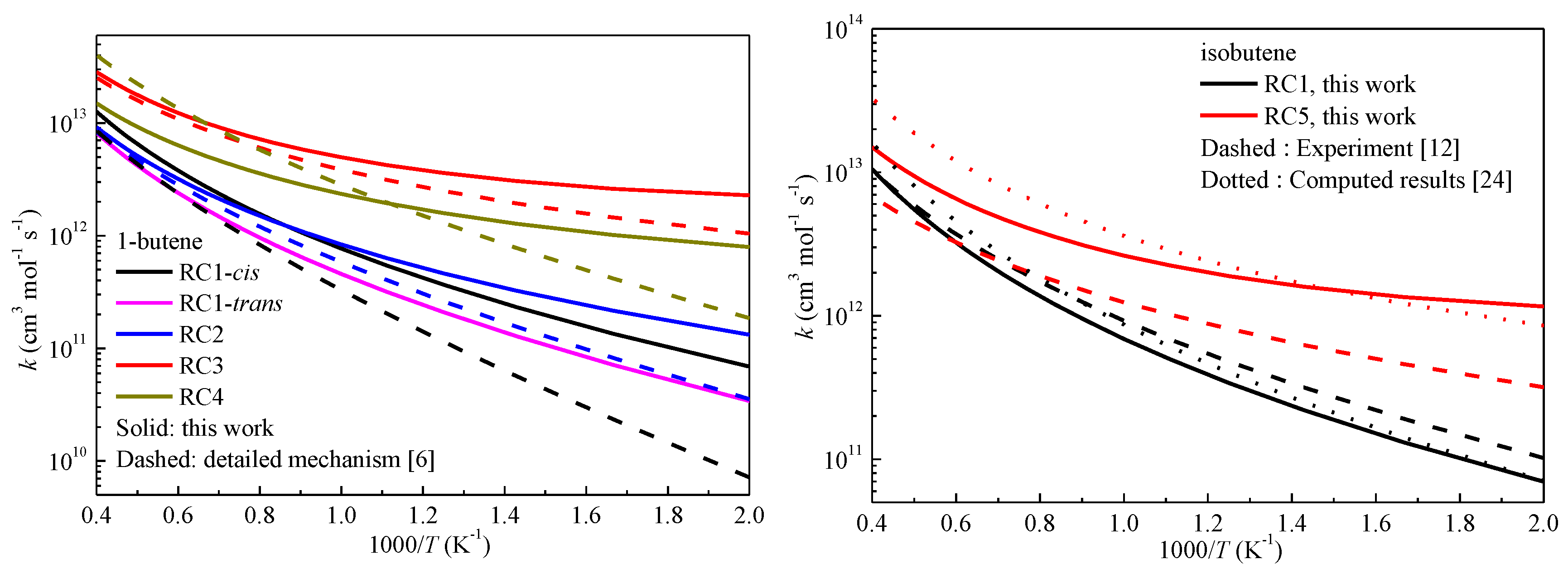
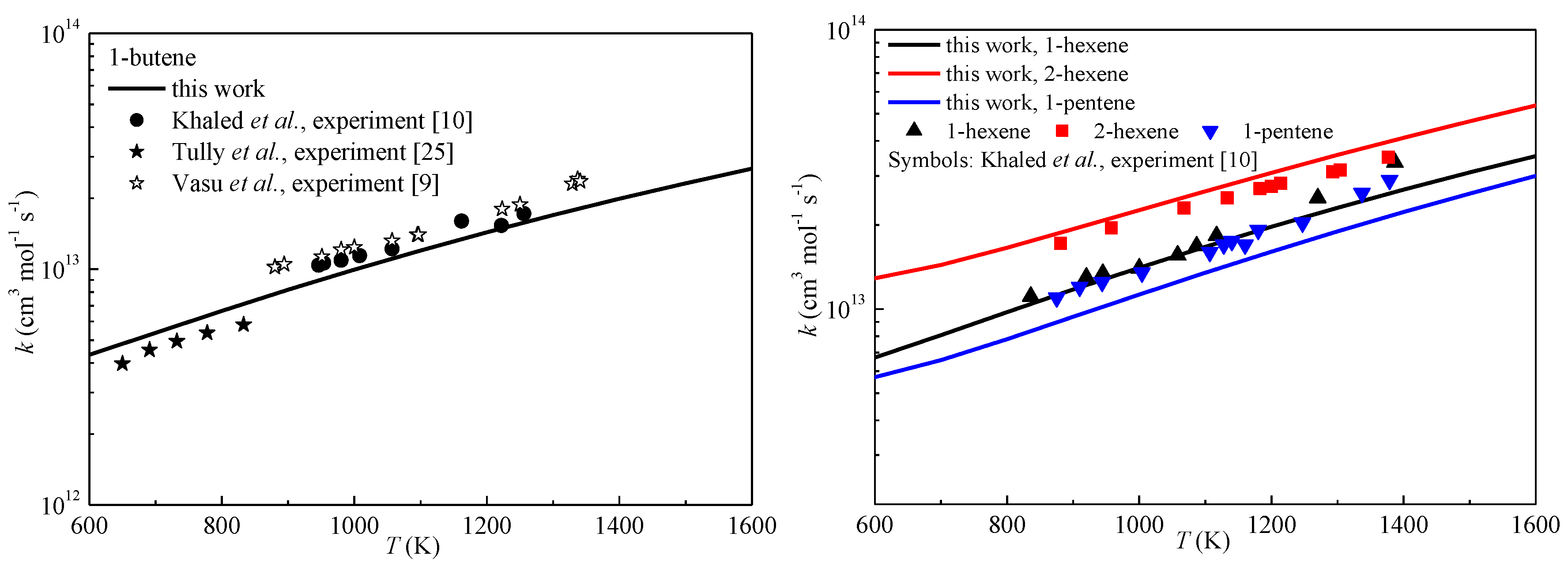
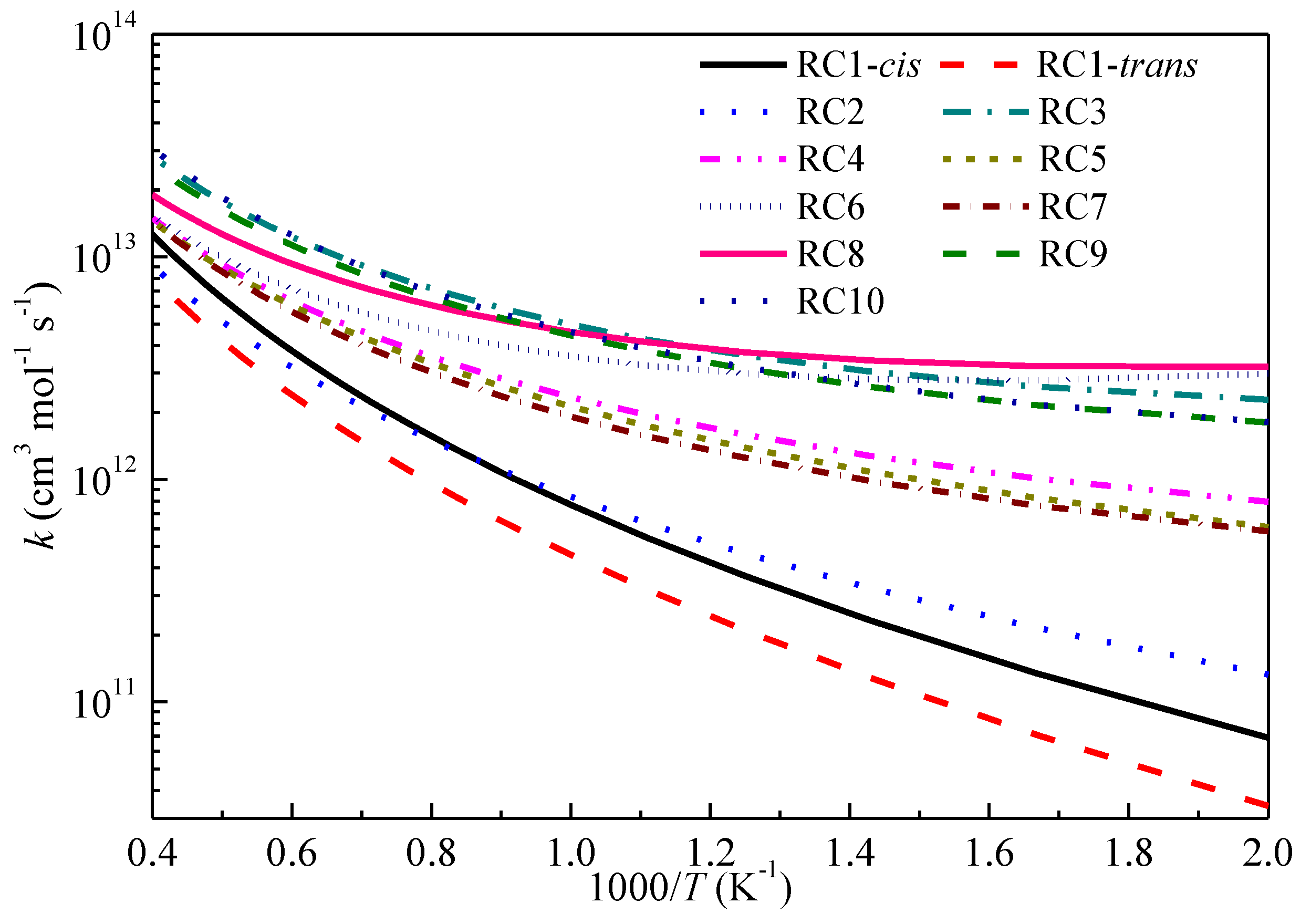
2.3. High-Pressure Limit Rate Constants
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jenkin, M.E.; Clemitshaw, K.C. Ozone and other secondary photochemical pollutants: Chemical processes governing their formation in the planetary boundary layer. Atmos. Environ. 2000, 34, 2499–2527. [Google Scholar] [CrossRef]
- Boot, M.D.; Tian, M.; Hensen, E.J.M.; Sarathy, S.M. Impact of fuel molecular structure on auto-ignition behavior-design rules for future high-performance gasolines. Prog. Energy Combust. Sci. 2017, 60, 1–25. [Google Scholar] [CrossRef]
- McGillen, M.R.; Percival, C.J.; Shallcross, D.E.; Harvey, J.N. Is hydrogen abstraction an important pathway in the reaction of alkenes with the OH radical? Phys. Chem. Chem. Phys. 2007, 9, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- Kopp, M.M.; Donato, N.S.; Petersen, E.L.; Metcalfe, W.K.; Burke, S.M.; Curran, H.J. Ignition and oxidation of ethylene-air mixtures at elevated pressures, Part 2: Chemical kinetics. J. Propul. Power 2014, 30, 799–811. [Google Scholar] [CrossRef]
- Wang, K.; Villano, S.M.; Dean, A.M. Fundamentally-based kinetic model for propene pyrolysis. Combust. Flame 2015, 162, 4456–4470. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, C.-W.; Somers, K.P.; Zhang, K.; Curran, H.J. The oxidation of 2-butene: A high pressure ignition delay, kinetic modeling study and reactivity comparison with isobutene and 1-butene. Proc. Combust. Inst. 2017, 36, 403–411. [Google Scholar] [CrossRef]
- Yang, F.; Deng, F.; Zhang, P.; Hu, E.; Cheng, Y.; Huang, Z. Comparative study on ignition characteristics of 1-hexene and 2-hexene behind reflected shock waves. Energy Fuels 2016, 30, 5130–5137. [Google Scholar] [CrossRef]
- Cheng, Y.; Hu, E.; Lu, X.; Li, X.; Gong, J.; Li, Q.; Huang, Z. Experimental and kinetic study of pentene isomers and n-pentane in laminar flames. Proc. Combust. Inst. 2017, 36, 1279–1286. [Google Scholar] [CrossRef]
- Vasu, S.S.; Huynh, L.K.; Davidson, D.F.; Hanson, R.K.; Golden, D.M. Reactions of OH with butene isomers: Measurements of the overall rates and a theoretical study. J. Phys. Chem. A 2011, 115, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- Khaled, F.; Badra, J.; Farooq, A. A shock tube study of C4-C6 straight chain alkenes + OH reactions. Proc. Combust. Inst. 2017, 36, 289–298. [Google Scholar] [CrossRef]
- Battin-Leclerc, F.; Blurock, E.; Bounaceur, R.; Fournet, R.; Glaude, P.A.; Herbinet, O.; Sirjean, B.; Warth, V. Towards cleaner combustion engines through groundbreaking detailed chemical kinetic models. Chem. Soc. Rev. 2011, 40, 4762–4782. [Google Scholar] [CrossRef] [Green Version]
- Khaled, F.; Giri, B.R.; Farooq, A. A high-temperature shock tube kinetic study for the branching ratios of isobutene + OH reaction. Proc. Combust. Inst. 2017, 36, 265–272. [Google Scholar] [CrossRef]
- Ratkiewicz, A.; Huynh, L.K.; Truong, T.N. Performance of first-principles-based reaction class transition state theory. J. Phys. Chem. B 2016, 120, 1871–1884. [Google Scholar] [CrossRef]
- Wang, Q.-D.; Liu, Z.-W. Reaction kinetics of hydrogen atom abstraction from C4−C6 alkenes by the hydrogen atom and methyl radical. J. Phys. Chem. A 2018, 122, 5202–5210. [Google Scholar] [CrossRef]
- Lee, T.J. Comparison of the T1 and D1 Diagnostics for electronic structure theory: A new definition for the open-shell D1 diagnostic. Chem. Phys. Lett. 2003, 372, 362–367. [Google Scholar] [CrossRef]
- Liu, G.; Ding, Y.; Li, Z.; Fu, Q.; Huang, X.; Sun, C.; Tang, A. Theoretical study on mechanisms of the high-temperature reactions C2H3+H2O and C2H4+OH. Phys. Chem. Chem. Phys. 2002, 4, 1021–1027. [Google Scholar] [CrossRef]
- Zador, J.; Jasper, A.W.; Miller, J.A. The reaction between propene and hydroxyl. Phys. Chem. Chem. Phys. 2009, 11, 11040–11053. [Google Scholar] [CrossRef]
- Huynh, L.K.; Zhang, H.R.; Zhang, S.; Eddings, E.; Sarofim, A.; Law, M.E.; Westmoreland, P.R.; Truong, T.N. Kinetics of enol formation from reaction of OH with propene. J. Phys. Chem. A 2009, 113, 3177–3185. [Google Scholar] [CrossRef]
- Izsak, R.; Szori, M.; Knowles, P.J.; Viskolcz, B. High accuracy ab initio calculations on reactions of OH with 1-alkenes. The case of propene. J. Chem. Theory Comput. 2009, 5, 2313–2321. [Google Scholar] [CrossRef]
- Sun, H.; Law, C.K. Kinetics of hydrogen abstraction reactions of butene isomers by OH radical. J. Phys. Chem. A 2010, 114, 12088–12098. [Google Scholar] [CrossRef]
- Greenwald, E.E.; North, S.W.; Georgievskii, Y.; Klippenstein, S.J. A two transition state model for radical-molecule reactions: A case study of the addition of OH to C2H4. J. Phys. Chem. A 2005, 109, 6031–6044. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Khaled, F.; Ning, H.; Ma, L.; Farooq, A.; Ren, W. Theoretical and shock tube study of the rate constants for hydrogen abstraction reactions of ethyl formate. J. Phys. Chem. A 2017, 121, 6304–6313. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.-D.; Ni, Z.-H. Theoretical and kinetic study of the hydrogen atom abstraction reactions of unsaturated C6 methyl esters with hydroxyl radical. Chem. Phys. Lett. 2016, 650, 119–125. [Google Scholar] [CrossRef]
- Zhou, C.-W.; Li, Y.; O’Connor, E.; Somers, K.P.; Thion, S.; Keesee, C.; Mathieu, O.; Petersen, E.L.; DeVerter, T.A.; Oehlschlaeger, M.A.; et al. A comprehensive experimental and modeling study of isobutene oxidation. Combust. Flame 2016, 167, 353–379. [Google Scholar] [CrossRef] [Green Version]
- Tully, F.P. Hydrogen-atom abstraction from alkenes by OH, ethene and 1-butene. Chem. Phys. Lett. 1998, 143, 510–514. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Goldsmith, C.F.; Green, W.H.; Klippenstein, S.J. Role of O2 + QOOH in low-temperature ignition of propane. 1. Temperature and pressure dependent rate coefficients. J. Phys. Chem. A 2012, 116, 3325–3346. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Canneaux, S.; Bohr, F.; Henon, E. KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results. J. Comput. Chem. 2014, 35, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Hirschfelder, J.O.; Wigner, E. Some quantum-mechanical considerations in the theory of reactions involving an activation energy. J. Chem. Phys. 1939, 7, 616. [Google Scholar] [CrossRef]
- McClurg, R.B.; Flagan, R.C.; Goddard, W.A., III. The hindered rotor density-of-states interpolation function. J. Chem. Phys. 1997, 106, 6675. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RC | Prototype Reaction | CCSD(T)-MP2/CBS | |
|---|---|---|---|
| ΔE | |||
| RC1-cis | CH2=CHCH2CH3 → •HC=CHCH2CH3 | 4.53 (4.35) | −7.58 (−7.69) |
| 6.01 (6.32) b | |||
| RC1-trans | CH2=CHCH2CH3 → •HC=CHCH2CH3 | 4.98 (4.78) | −7.10 (−7.23) |
| 6.04 b | |||
| RC2 | CH2=CHCH2CH3 → CH2=C•CH2CH3 | 2.96 (2.78) | −10.99 (−11.13) |
| 3.90 (3.79) b | |||
| RC3 | CH2=CHCH2CH3 → CH2=CHCH•CH3 | 0.27 (0.09) | −34.08 (−34.17) |
| −0.01 (−0.41) b | |||
| RC4 | CH2=CHCH2CH3 → CH2=CHCH2CH2• | 1.43 (1.27) | −17.57 (−17.52) |
| 2.27 (2.40) b | |||
| RC5 | CH3CH=CHCH3 → CH3CH=CHCH2• | 1.40 | −31.33 |
| RC6 | CH2=CHCH2CH2CH3 → CH=CHCH2CH•CH3 | −0.71 | −20.52 |
| RC7 | CH2=CHCH2CH2CH3 → CH=CHCH2CH2CH2• | 1.98 | −17.98 |
| RC8 | H2C=CHCH(CH3)2 → H2C=CHC•(CH3)2 | −0.59 | −35.59 |
| RC9 | CH2=CH(CH2)2CH2CH3 → CH2=CH(CH2)2CH•CH3 | 0.68 | −20.45 |
| RC10 | CH2=CHCH2CH=CH2 → CH2=CHCH•CH=CH2 | 0.19 | −42.10 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.-D.; Sun, M.-M.; Liang, J.-H. Reaction Mechanisms and Kinetics of the Hydrogen Abstraction Reactions of C4–C6 Alkenes with Hydroxyl Radical: A Theoretical Exploration. Int. J. Mol. Sci. 2019, 20, 1275. https://doi.org/10.3390/ijms20061275
Wang Q-D, Sun M-M, Liang J-H. Reaction Mechanisms and Kinetics of the Hydrogen Abstraction Reactions of C4–C6 Alkenes with Hydroxyl Radical: A Theoretical Exploration. International Journal of Molecular Sciences. 2019; 20(6):1275. https://doi.org/10.3390/ijms20061275
Chicago/Turabian StyleWang, Quan-De, Mao-Mao Sun, and Jin-Hu Liang. 2019. "Reaction Mechanisms and Kinetics of the Hydrogen Abstraction Reactions of C4–C6 Alkenes with Hydroxyl Radical: A Theoretical Exploration" International Journal of Molecular Sciences 20, no. 6: 1275. https://doi.org/10.3390/ijms20061275
APA StyleWang, Q.-D., Sun, M.-M., & Liang, J.-H. (2019). Reaction Mechanisms and Kinetics of the Hydrogen Abstraction Reactions of C4–C6 Alkenes with Hydroxyl Radical: A Theoretical Exploration. International Journal of Molecular Sciences, 20(6), 1275. https://doi.org/10.3390/ijms20061275