In Silico Analysis of Homologous Heterodimers of Cruzipain-Chagasin from Structural Models Built by Homology

 ,
,  , ,
, ,
Abstract
:
1. Introduction
2. Results
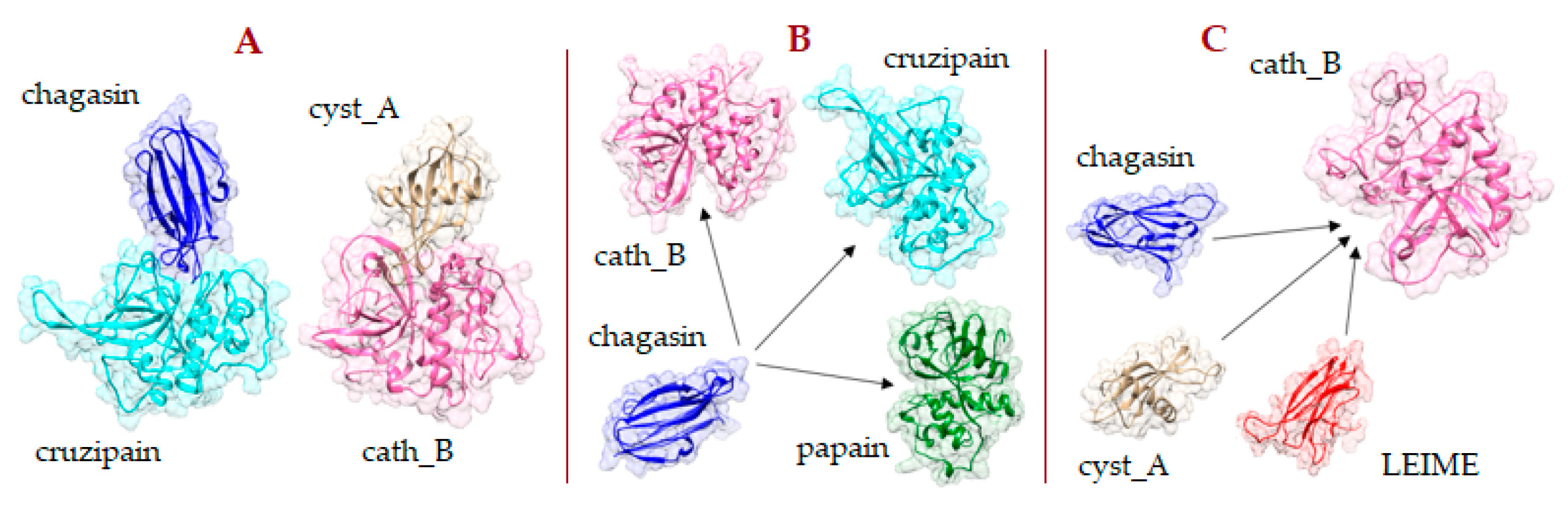
2.1. Heterodimer Models Constructed by Homology Modeling
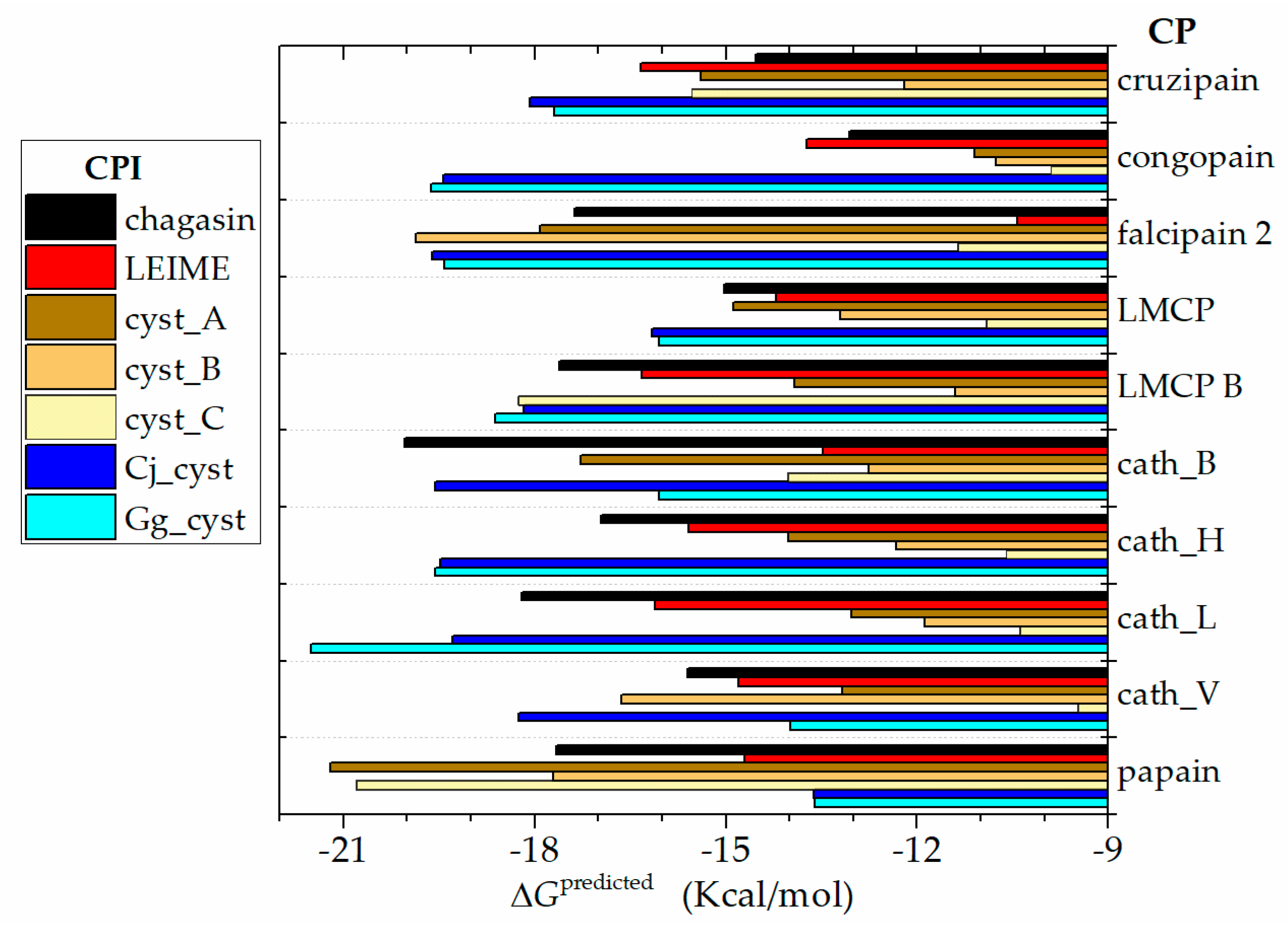
2.2. ΔGb Prediction of Homologs of the Cruzipain−Chagasin Complex
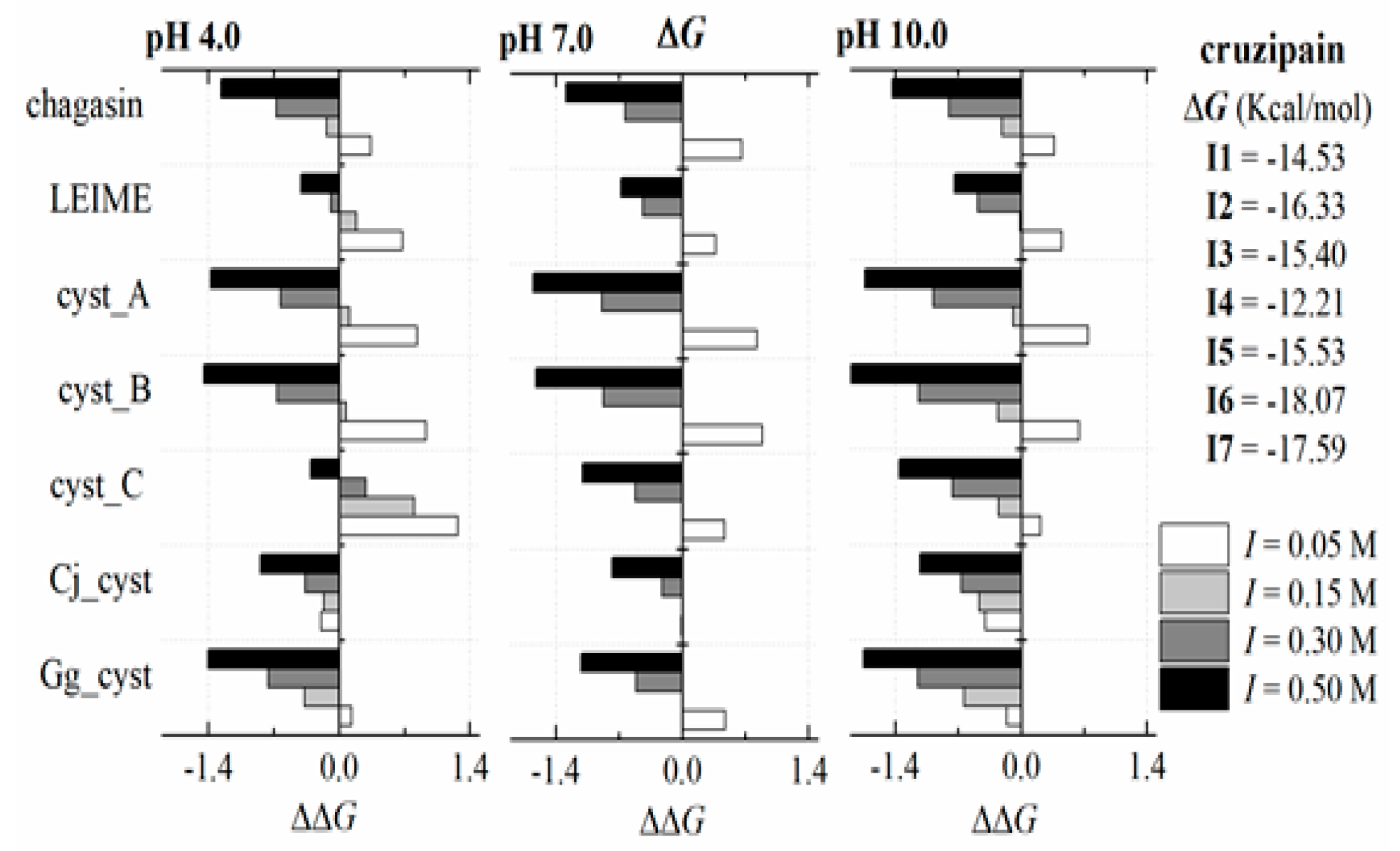
2.3. Energetic Analysis
3. Discussion
3.1. Analysis of Ki Values Obtained in In Vitro Experiments (Reported in the Literature)
3.2. Molecular Models Constructed by Homology Modeling and Methodology Validation
3.3. Analysis of ΔGb Prediction Values
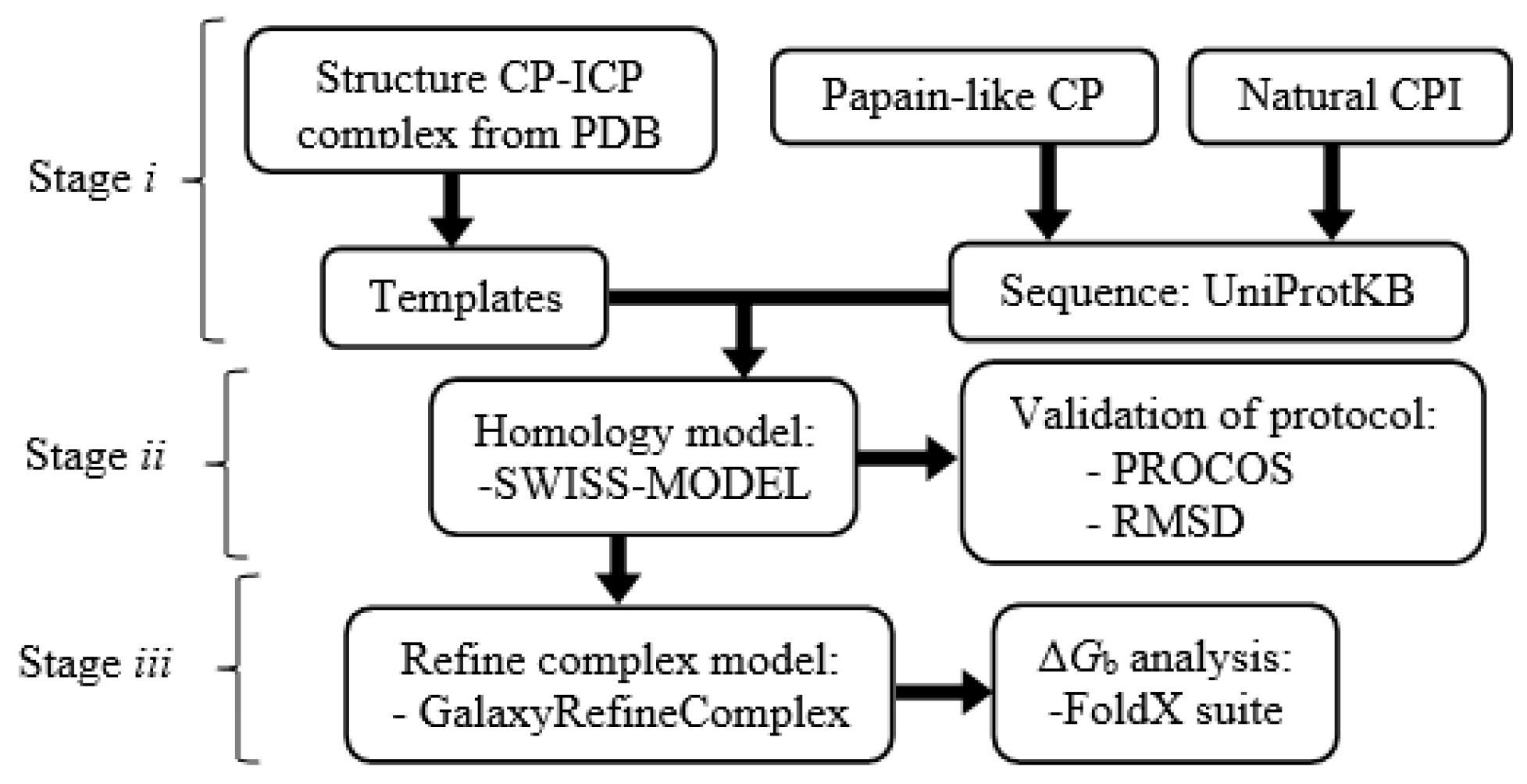
4. Materials and Methods
4.1. Preparation and Analysis of Sequences
4.2. Structural Modeling by Homology
4.3. Evaluation of Heterodimer Models and ΔGb Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ΔGb | binding energy |
| CP | cysteine proteinase |
| ICP | cysteine protease inhibitors |
| cath | cathepsin |
| QMQE | the global model quality estimation |
| cyst | ciystatin |
| I | ionic strength |
Appendix A
Residues Present in the Interaction of CP to Natural ICP from Crystal Structures

Appendix B
Appendix B.1. Contribution of ΔH y ΔS to ΔG

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ICP | chagasin | LEIME | cyst_A | cyst_B | cyst_C | Cj_cyst | Gg_cyst | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CP | ΔH | −TΔS | ΔH | −TΔS | ΔH | −TΔS | ΔH | −TΔS | ΔH | −TΔS | ΔH | −TΔS | ΔH | −TΔS |
| E1 | −20.3 | 5.8 | −17.0 | 0.6 | −19.0 | 3.5 | −23.2 | 11.0 | −18.5 | 2.9 | −22.6 | 4.5 | −22.8 | 5.1 |
| E2 | −16.6 | 3.6 | −15.3 | 1.5 | −18.4 | 7.3 | −18.3 | 7.5 | −12.5 | 2.6 | −28.2 | 8.7 | −26.8 | 7.2 |
| E3 | −22.1 | 4.8 | −10.8 | 0.4 | −20.2 | 2.3 | −27.9 | 8.1 | −13.3 | 2.0 | −21.2 | 1.5 | −20.9 | 1.5 |
| E4 | −21.7 | 6.7 | −16.5 | 2.4 | −24.1 | 9.2 | −21.7 | 8.5 | −15.7 | 4.8 | −23.8 | 7.7 | −23.5 | 7.5 |
| E5 | −18.1 | 0.5 | −17.3 | 1.0 | −21.6 | 7.7 | −21.5 | 10.1 | −23.7 | 5.5 | −21.9 | 3.7 | −22.0 | 3.4 |
| E6 | −21.6 | 1.5 | −15.7 | 2.3 | −22.0 | 4.8 | −21.7 | 8.9 | −16.4 | 2.4 | −23.2 | 3.6 | −23.0 | 7.0 |
| E7 | −22.9 | 5.9 | −22.8 | 7.3 | −22.5 | 8.4 | −19.5 | 7.1 | −14.0 | 3.4 | −28.8 | 9.3 | −23.9 | 4.3 |
| E8 | −19.8 | 1.6 | −18.1 | 1.9 | −24.5 | 11.4 | −26.4 | 14.6 | −14.6 | 4.3 | −34.8 | 15.6 | −31.8 | 10.2 |
| E9 | −19.8 | 4.2 | −19.4 | 4.6 | −26.3 | 13.1 | −22.9 | 6.2 | −13.1 | 3.6 | −25.8 | 7.6 | −27.7 | 8.4 |
| E10 | −22.8 | 5.1 | −21.2 | 6.5 | −30.0 | 8.7 | −24.1 | 6.4 | −26.5 | 5.7 | −21.1 | 7.5 | −22.6 | 9.0 |
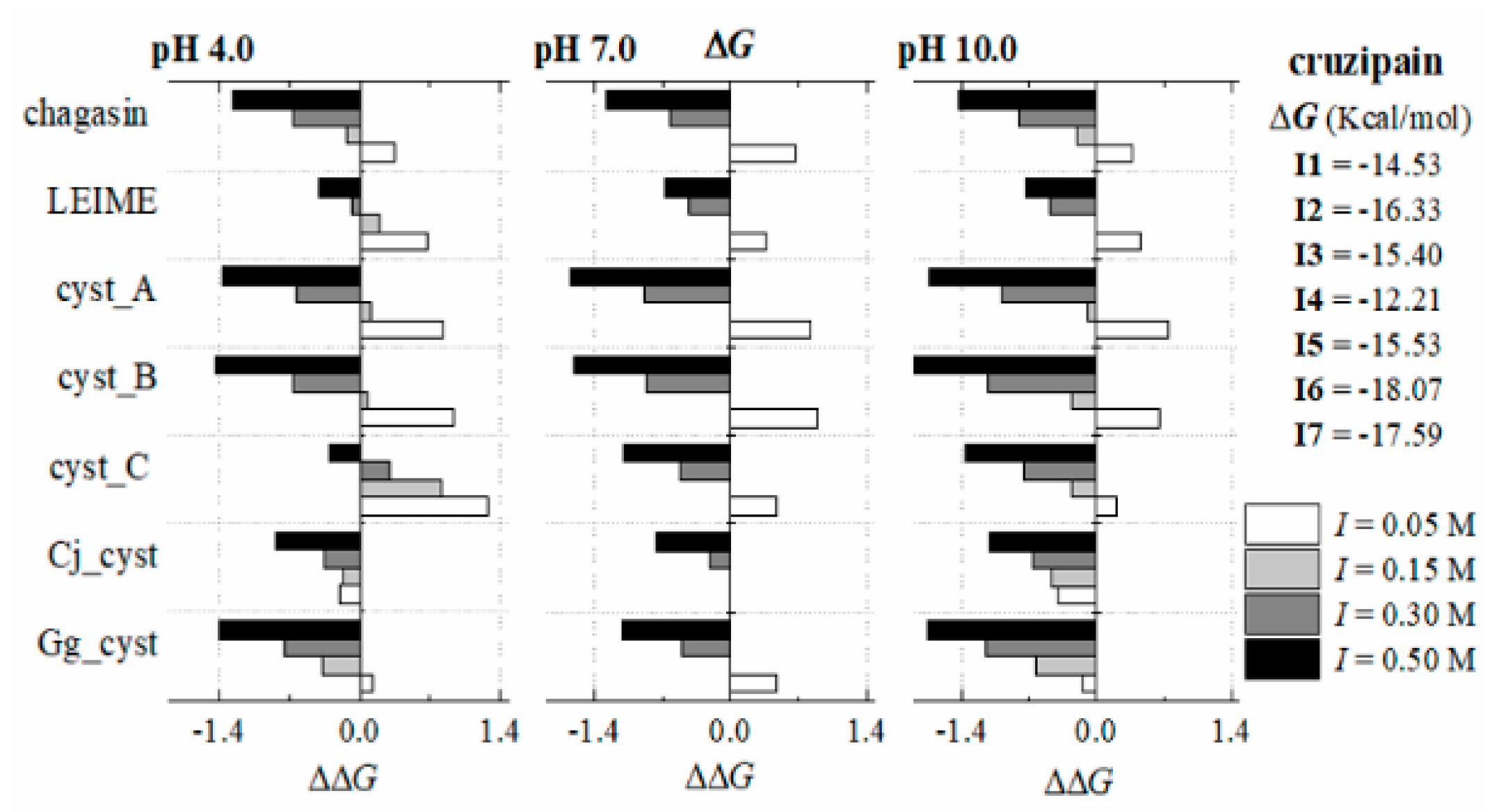
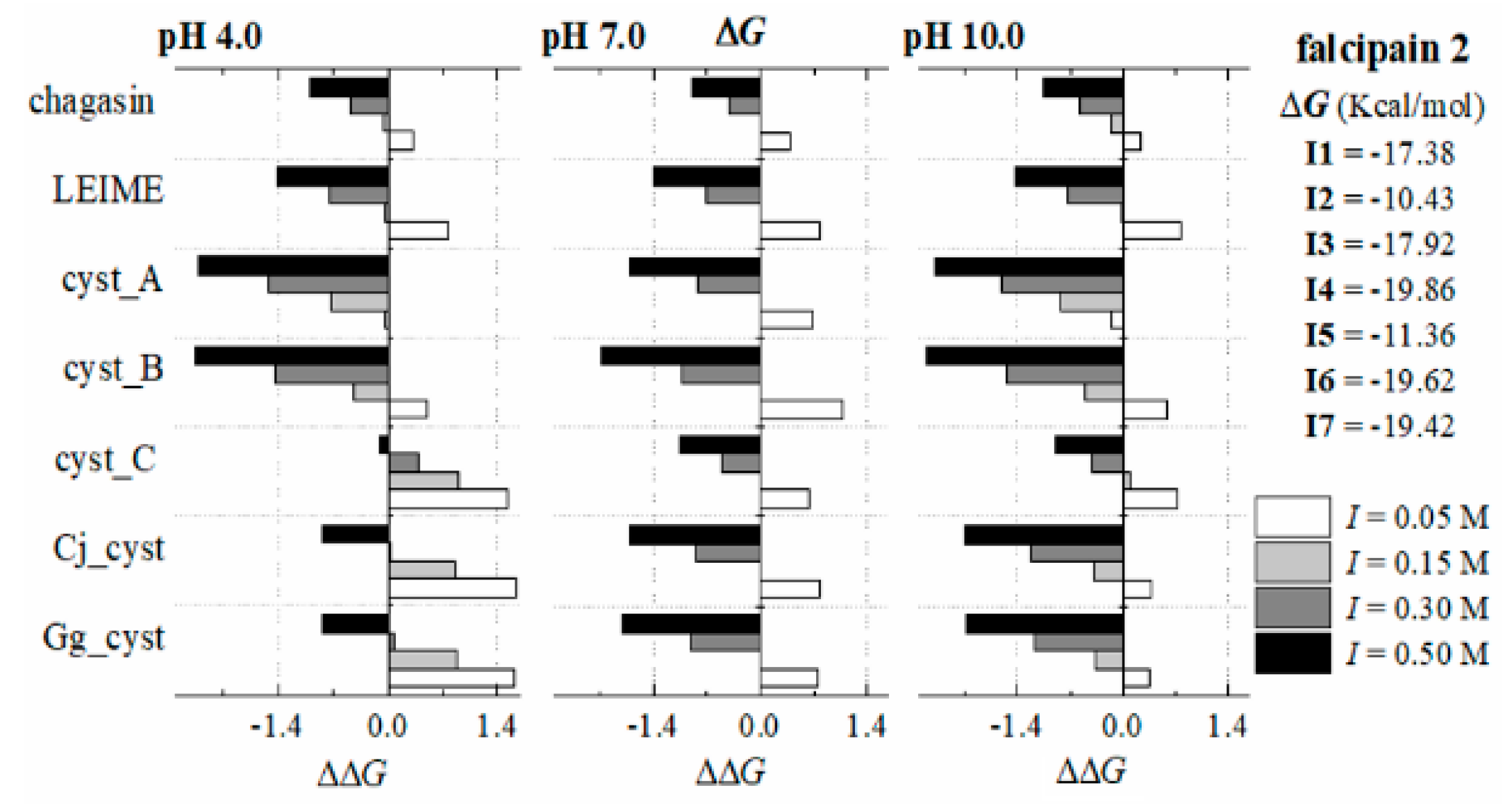
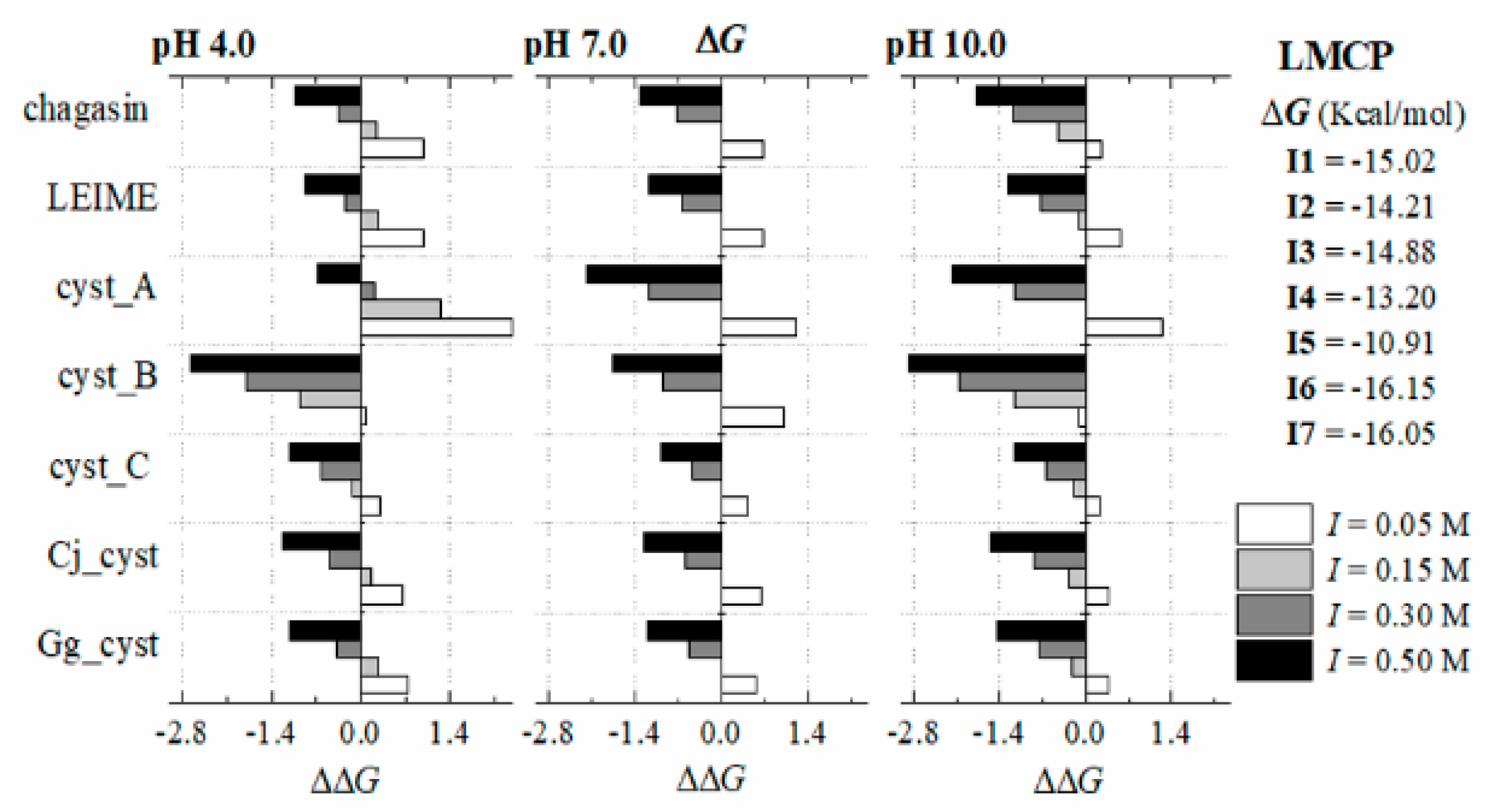
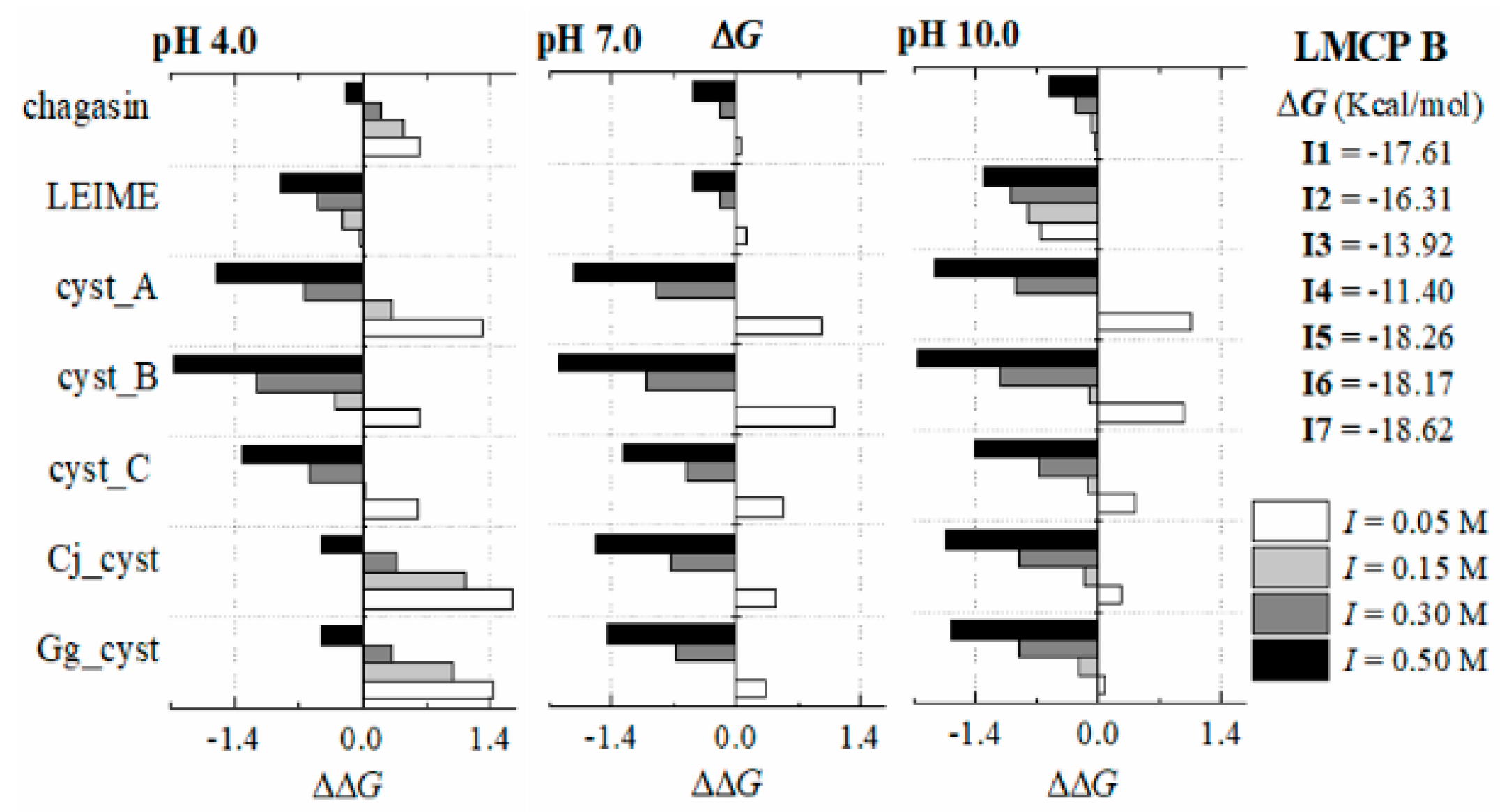
Appendix B.2. Dependence of ΔGb (I, pH) at 298 K

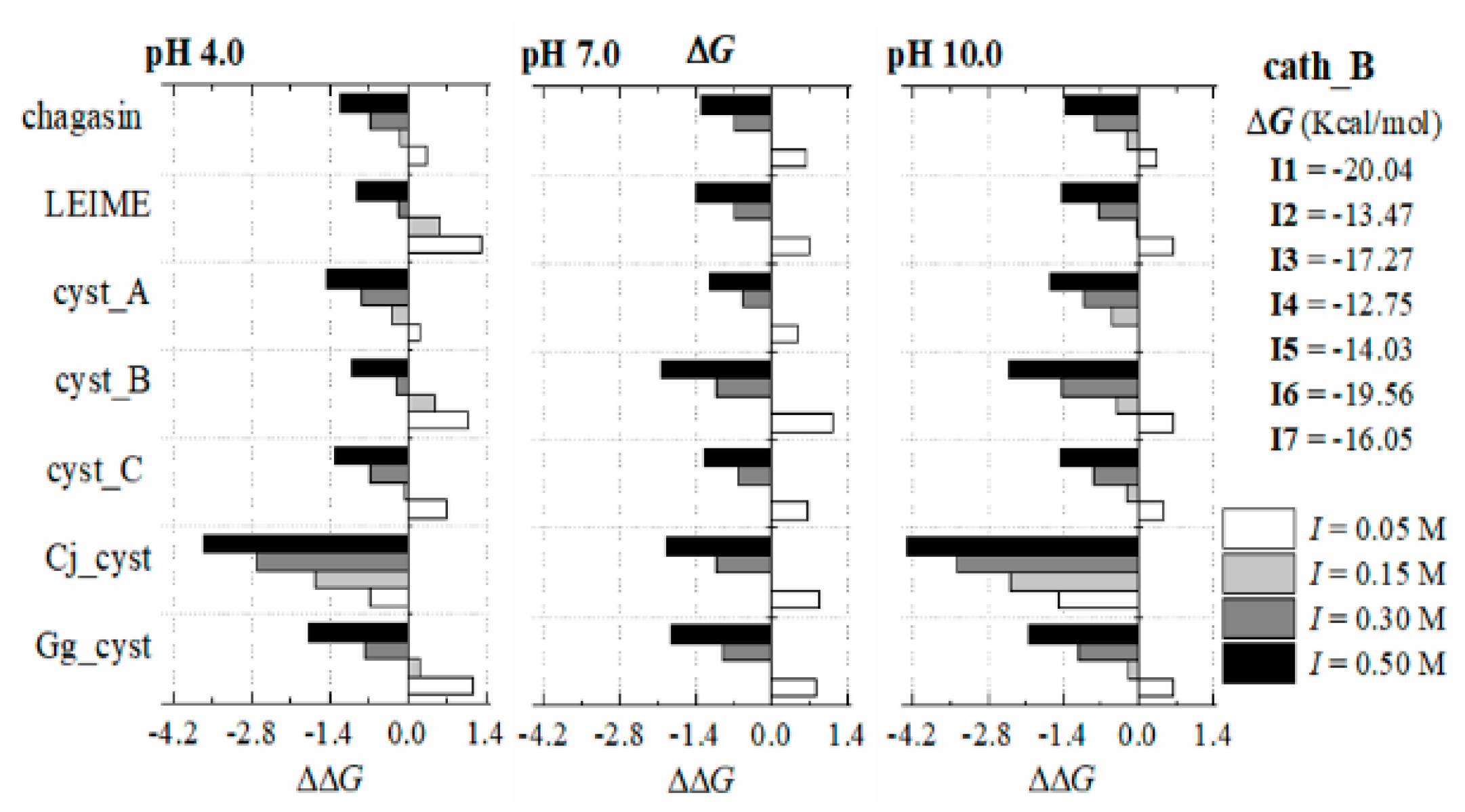
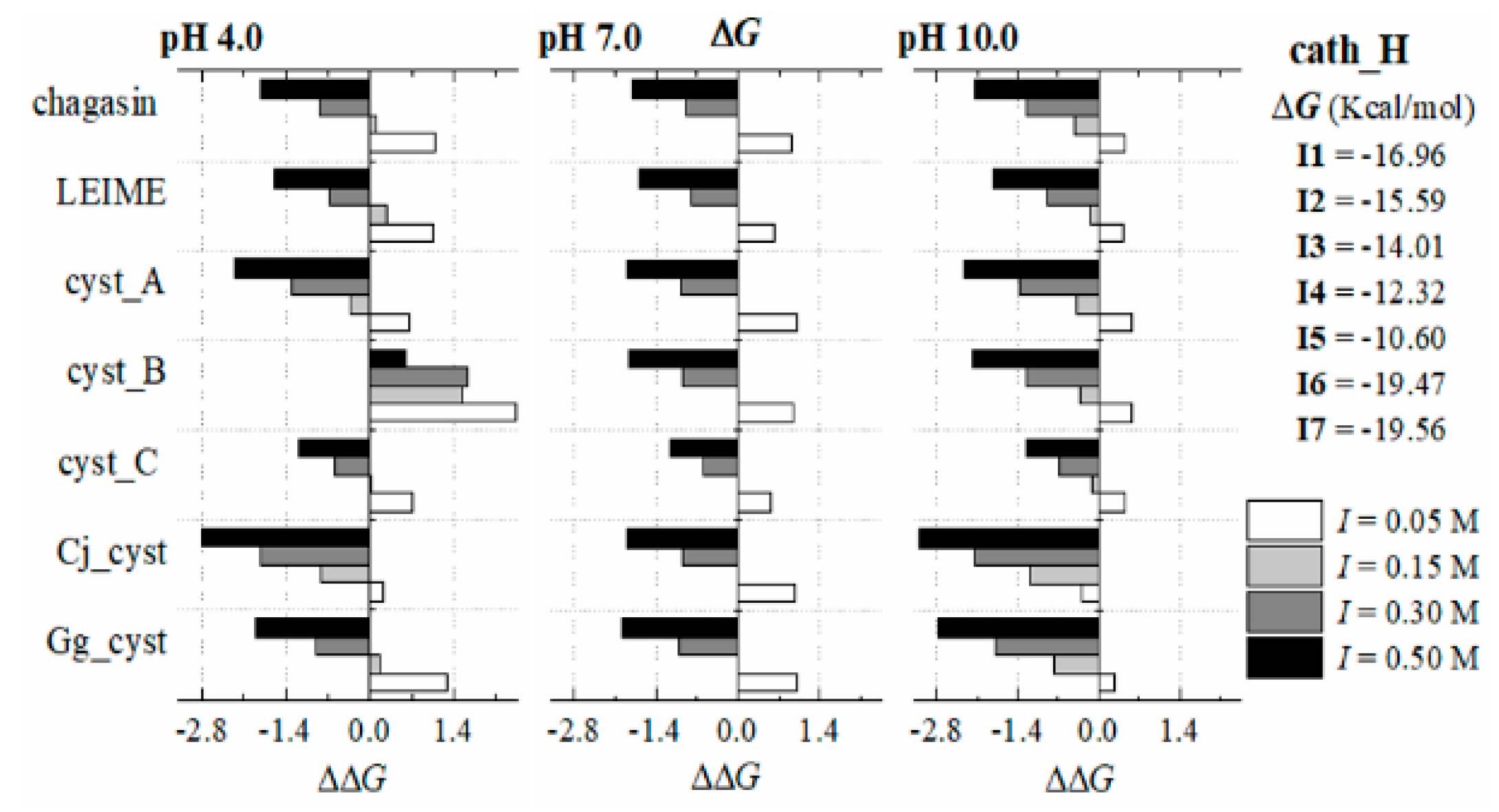
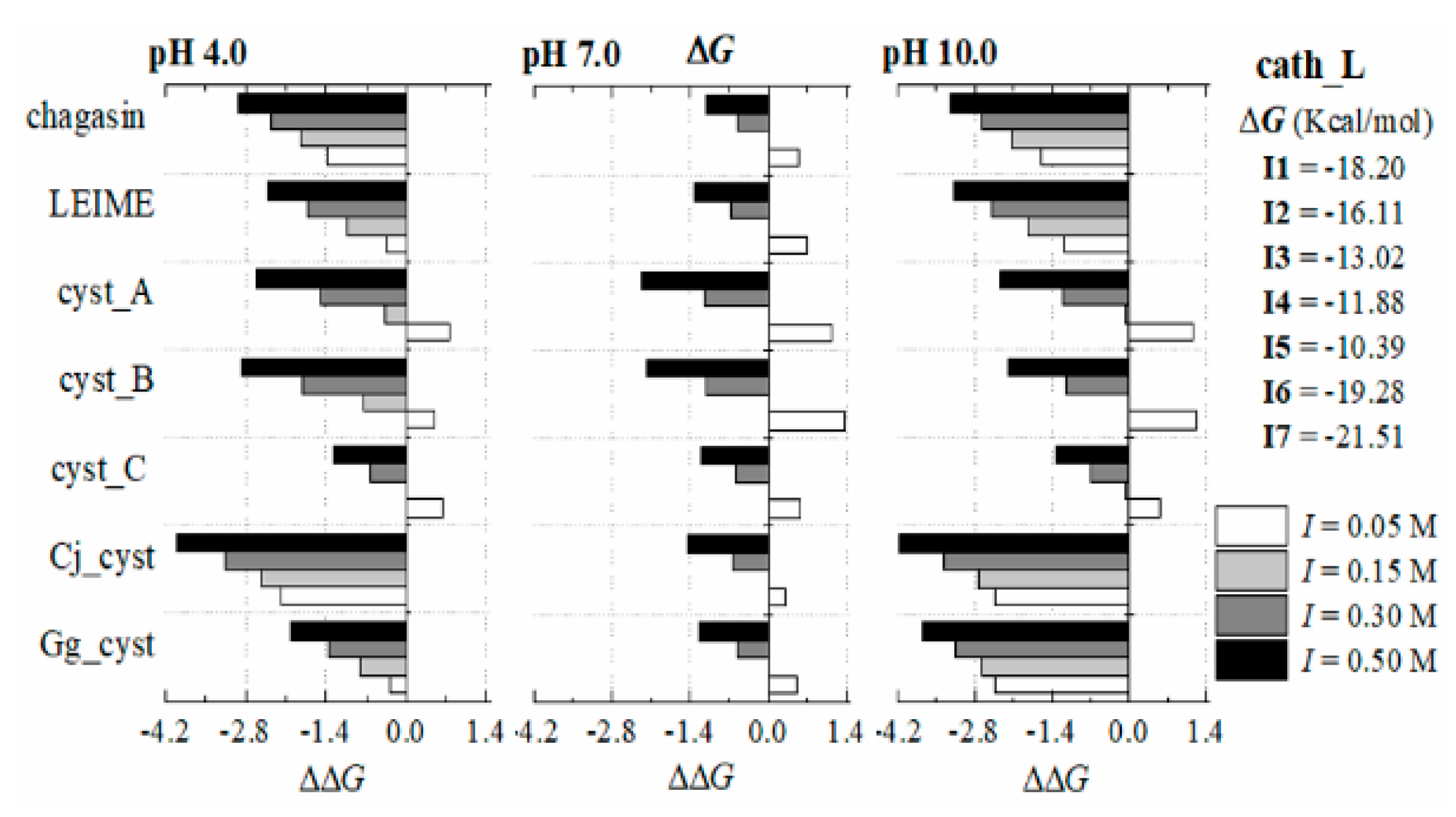
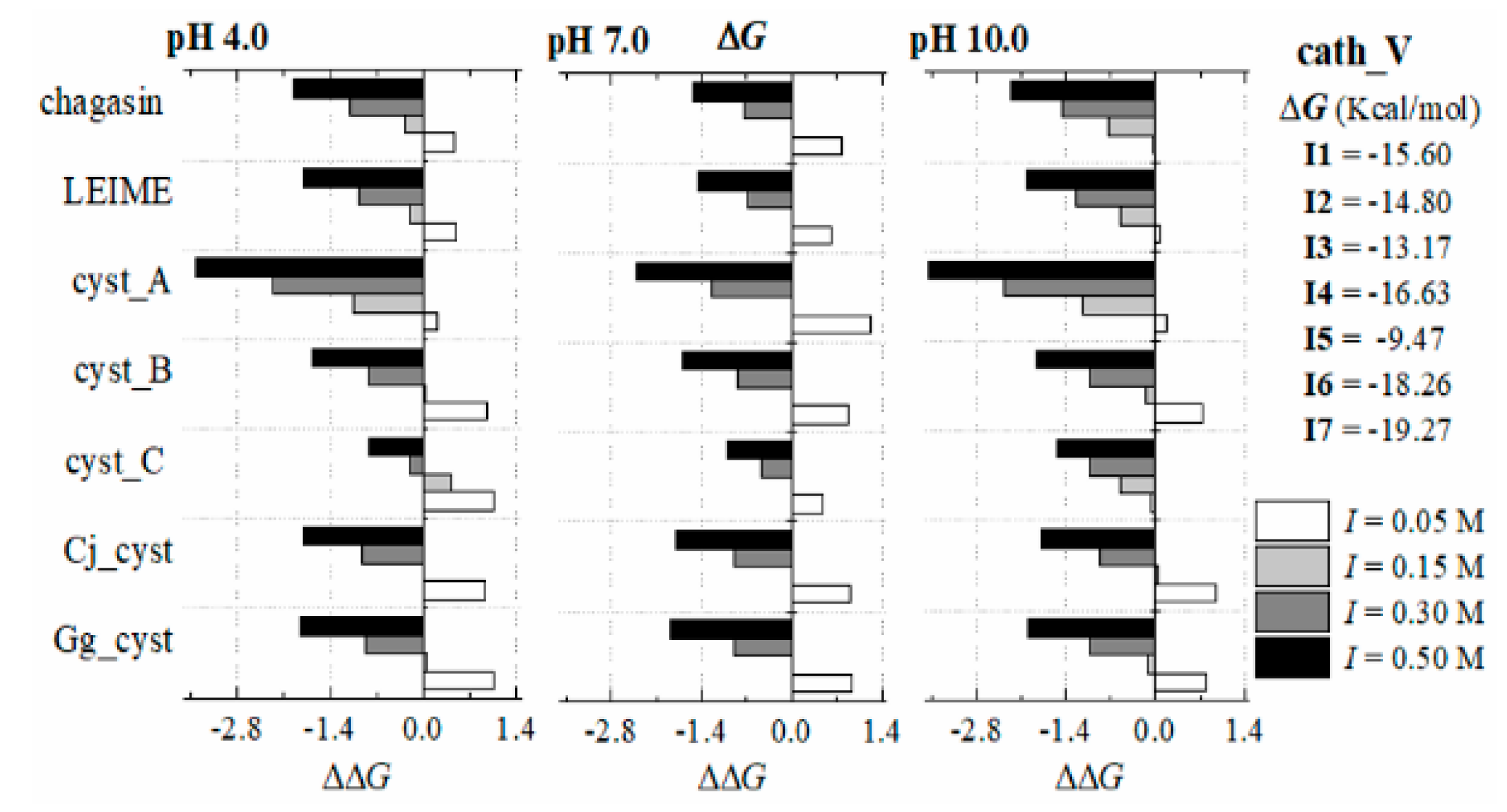

References
- Sueth-Santiago, V.; Decote-Ricardo, D.; Morrot, A.; Freire-de-Lima, C.G.; Freire Lima, M.E. Challenges in the chemotherapy of Chagas disease: Looking for possibilities related to the differences and similarities between the parasite and host. World J. Biol. Chem. 2017, 26, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Caballero, C.I.; Dzul-Rosado, K.; Dzul-Tut, I.; Balam-May, I.; Zavala-Castro, J. Knowledge of vector-borne diseases (dengue, rickettsiosis and Chagas disease) in physicians. Gaceta Médica de México 2017, 153, 321–328. [Google Scholar] [PubMed]
- Liu, Q.; Zhou, X.N. Preventing the transmission of American trypanosomiasis and its spread into non-endemic countries. Infect. Dis. Poverty 2015, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Roca, C.S.; Soriano-Arandes, A.; Solsona, D.L.; Gascón, B.J. Grupo de consenso Chagas-APS. Documento de consenso sobre el abordaje de la enfermedad de Chagas en atención primaria de salud de áreas no endémicas. Atención Primaria 2015, 475, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Mottram, J.C.; Helms, M.J.; Coombs, G.H.; Sajid, M. Clan CD cysteine peptidases of parasitic protozoa. TRENDS Parasitol. 2003, 19, 182–187. [Google Scholar] [CrossRef]
- Joon-Yong, C.; Young-An, B.; Byoung-Kuk, N.; Yoon, K. Cysteine protease inhibitors as potential antiparasitic agents. Expert Opin. Ther. Pat. 2005, 15, 995–1007. [Google Scholar] [CrossRef]
- Van Wyk, S.G.; Kunert, K.J.; Cullis, C.A.; Pillay, P.; Makgopa, M.E.; Schlüter, U.; Vorster, B.J. Review: The future of cystatin engineering. Plant Sci. 2016, 246, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Leto, G.; Crescimanno, M.; Flandina, C. On the role of cystatin C in cancer progression. Life Sci. 2018, 202, 152–160. [Google Scholar] [CrossRef]
- Costa, F.R.; Lima Ana, C.A. Natural cysteine protease inhibitors in protozoa: Fifteen years of the chagasin family. Biochimie 2016, 122, 197–207. [Google Scholar] [CrossRef]
- Monteiro, A.C.; Abrahamson, M.; Lima, A.P.; Vannier-Santos, M.A.; Scharfstein, J. Identification, characterization and localization of chagasin, a tight-binding cysteine protease inhibitor in Trypanosoma cruzi. J. Cell Sci. 2002, 114, 3933–3942. [Google Scholar]
- Smith, B.O.; Picken, N.C.; Westrop, G.D.; Bromek, K.; Mottram, J.C.; Coombs, G.H. The structure of leishmania mexicana ICP provides evidence for convergent evolution of cysteine peptidase inhibitors. J. Biol. Chem. 2006, 281, 5821–5828. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Nair, S.; Sen, N.; Soni, N.; Madhusudhant, M.S. In silico methods for design of biological therapeutics. Methods 2017, 131, 33–65. [Google Scholar] [CrossRef] [PubMed]
- Dourado, D.F.; Flores, S.C. Modeling and fitting protein-protein complexes to predict change of binding energy. Sci. Rep. 2016, 6, 25406. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- França, T.C. Homology modeling: An important tool for the drug discovery. J. Biomol. Struct. Dyn. 2015, 33, 1780–1793. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; D’Ursi, P.; Moscatelli, M.; Bruno, O.; Orro, A.; Rotolo, C.; Milanesi, L.; Fossa, P. Homology modeling, docking studies and molecular dynamic simulations using graphical processing unit architecture to probe the type-11 phosphodiesterase catalytic site: A computational approach for the rational design of selective inhibitors. Chem. Biol. Drug 2013, 82, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Moro, S.; Deflorian, F.; Bacilieri, M.; Spalluto, G. Ligand-based homology modeling as attractive tool to inspect GPCR structural plasticity. Curr. Pharm. Des. 2006, 12, 2175–2185. [Google Scholar] [CrossRef]
- Cichero, E.; Espinoza, S.; Tonelli, M.; Franchini, S.; Gerasimo, A.S.; Sorbi, C.; Gainetdinov, R.R.; Brasili, L.; Fossa, P. A homology modelling-driven study leading to the discovery of the first mouse trace amine-associated receptor 5 (TAAR5) antagonists. Med. Chem. Commun. 2016, 7, 353–364. [Google Scholar] [CrossRef]
- Tastan Bishop, O.; Kroon, M. Study of protein complexes via homology modeling, applied to cysteine proteases and their protein inhibitors. J. Mol. Model. 2011, 17, 3163–3172. [Google Scholar] [CrossRef] [PubMed]
- Tyzack, J.D.; Furnham, N.; Sillitoe, I.; Orengo, C.M.; Thornton, J.M. Understanding enzyme function evolution from a computational perspective. Curr. Opin. Struct. Biol. 2017, 47, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.; Martins, M.; Jing, S.; Fu, J.; Cavaco-Paulo, A. Practical insights on enzyme stabilization. Crit. Rev. Biotechnol. 2018, 38, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Van Durme, J.; Delgado, J.; Stricher, F.; Serrano, L.; Schymkowitz, J.; Rousseau, F. A graphical interface for the FoldX force field. Bioinformatics 2011, 27, 1711–1712. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redzynia, I.; Ljunggren, A.; Bujacz, A.; Abrahamson, M.; Jaskolski, M.; Bujacz, G. Crystal structure of the parasite inhibitor chagasin in complex with papain allows identification of structural requirements for broad reactivity and specificity determinants for target proteases. FEBS J. 2009, 276, 793–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.X.; Pandey, K.C.; Scharfstein, J.; Whisstock, J.; Huang, R.K.; Jacobelli, J.; Fletterick, R.J.; Rosenthal, P.J.; Abrahamson, M.; Brinen, L.S.; et al. The structure of chagasin in complex with a cysteine protease clarifies the binding mode and evolution of an inhibitor family. Structure 2007, 15, 535–543. [Google Scholar] [CrossRef]
- Ljunggren, A.; Redzynia, I.; Alvarez-Fernandez, M.; Abrahamson, M.; Mort, J.S.; Krupa, J.C.; Jaskolski, M.; Bujacz, G. Crystal structure of the parasite protease inhibitor chagasin in complex with a host target cysteine protease. J. Mol. Biol. 2007, 371, 137–153. [Google Scholar] [CrossRef]
- Rowan, A.D.; Brzin, J.; Buttle, D.J.; Barrett, A.J. Inhibition of cysteine proteinases by a protein inhibitor from patato. FEBS Lett. 1990, 269, 328–330. [Google Scholar] [CrossRef]
- Chagas, J.R.; Authie, E.; Serveau, C.; Lalmanach, G.; Juliano, L.; Gauthier, F. A comparison of the enzymatic properties of the major cysteine proteinases from Trypanosoma congolense and Trypanosoma cruzi. Mol. Biochem. Parasitol. 1997, 88, 85–94. [Google Scholar] [CrossRef]
- Gerhartz, B.; Engh, R.A.; Mentele, R.; Eckerskorn, C.; Torquato, R.; Wittmann, J.; Kolb, H.J.; Machleidt, W.; Fritz, H.; Auerswald, E.A. Quail cystatin: Isolation and characterisation of a new member of the cystatin family and its hypothetical interaction with cathepsin B. FEBS Lett. 1997, 412, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Lima, A.P.; dos Reis, F.C.; Serveau, C.; Lalmanach, G.; Juliano, L.; Ménard, R.; Vernet, T.; Thomas, D.Y.; Storer, A.C.; Scharfstein, J. Cysteine protease isoforms from Trypanosoma cruzi, cruzipain 2 and cruzipain, present different substrate preference and susceptibility to inhibitors. Mol. Biochem. Parasitol. 2001, 114, 41–52. [Google Scholar] [CrossRef]
- Dos Reis, F.C.; Smith, B.O.; Santos, C.C.; Costa, T.F.; Scharfstein, J.; Coombs, G.H.; Mottram, J.C.; Lima, A.P. The role of conserved residues of chagasin in the inhibition of cysteine peptidases. FEBS Lett. 2008, 582, 485–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoka, V.; Nycander, M.; Lenarčič, B.; Labriola, C.; Cazzulo, J.J.; Björk, I.; Turk, V. Inhibition of cruzipain, the major cysteine proteinase of the protozoan parasite, Trypanosoma cruzi, by proteinase inhibitors of the cystatin superfamily. FEBS Lett. 1995, 370, 101–104. [Google Scholar] [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [Google Scholar] [CrossRef] [PubMed]
- Fink, F.; Hochrein, J.; Wolowski, V.; Merkl, R.; Gronwald, W. PROCOS: Computational analysis of protein–protein complexes. J. Comput. Chem. 2011, 32, 2575–2586. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Lee, H.; Seok, C. GalaxyRefineComplex: Refinement of protein-protein complex model structures driven by interface repacking. Sci. Rep. 2016, 6, 32153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Reyes-Espinosa, F.; Arroyo-Reyna, A.; García-Gutiérrez, P.; Serratos, I.N.; Zubillaga, R.A. Effects of pH on the association between the inhibitor cystatin and the proteinase chymopapain. Protein Pept. Lett. 2015, 22, 239–247. [Google Scholar] [CrossRef]
- Kakkar, T.; Boxenbaum, H.; Mayersohn, M. Estimation of Ki in a competitive enzyme-inhibition model: Comparisons among three methods of data analysis. Drug Metab. Dispos. 1999, 27, 756–762. [Google Scholar]
- Borea, P.A.; Varani, K.; Gessi, S.; Gilli, P.; Dalpiaz, A. Receptor binding thermodynamics as a tool for linking drug efficacy and affinity. Farmaco 1998, 53, 249–254. [Google Scholar] [CrossRef]
- Krieger, E.; Joo, K.; Lee, J.; Raman, S.; Thompson, J.; Tyka, M.; Baker, D.; Karplus, K. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins 2009, 77, 114–122. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Buß, O.; Rudat, J.; Ochsenreither, K. FoldX as protein engineering tool: Better than random based approaches? Comput. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar] [CrossRef]
- The FoldX Suite. Available online: URL http://foldxsuite.crg.eu/products (accessed on 1 February 2019).
- Shoemaker, B.A.; Zhang, D.; Tyagi, M.; Thangudu, R.R.; Fong, J.H.; Marchler-Bauer, A.; Bryant, S.H.; Madej, T.; Panchenko, A.R. IBIS (Inferred Biomolecular Interaction Server) reports, predicts and integrates multiple types of conserved interactions for proteins. Nucleic Acids Res. 2012, 40, D834–D840. [Google Scholar] [CrossRef]
- Shoemaker, B.A.; Zhang, D.; Thangudu, R.R.; Tyagi, M.; Fong, J.H.; Marchler-Bauer, A.; Bryant, S.H.; Madej, T.; Panchenko, A.R. Inferred Biomolecular Interaction Server—A web server to analyze and predict protein interacting partners and binding sites. Nucleic Acids Res. 2010, 38, D518–D524. [Google Scholar] [CrossRef]
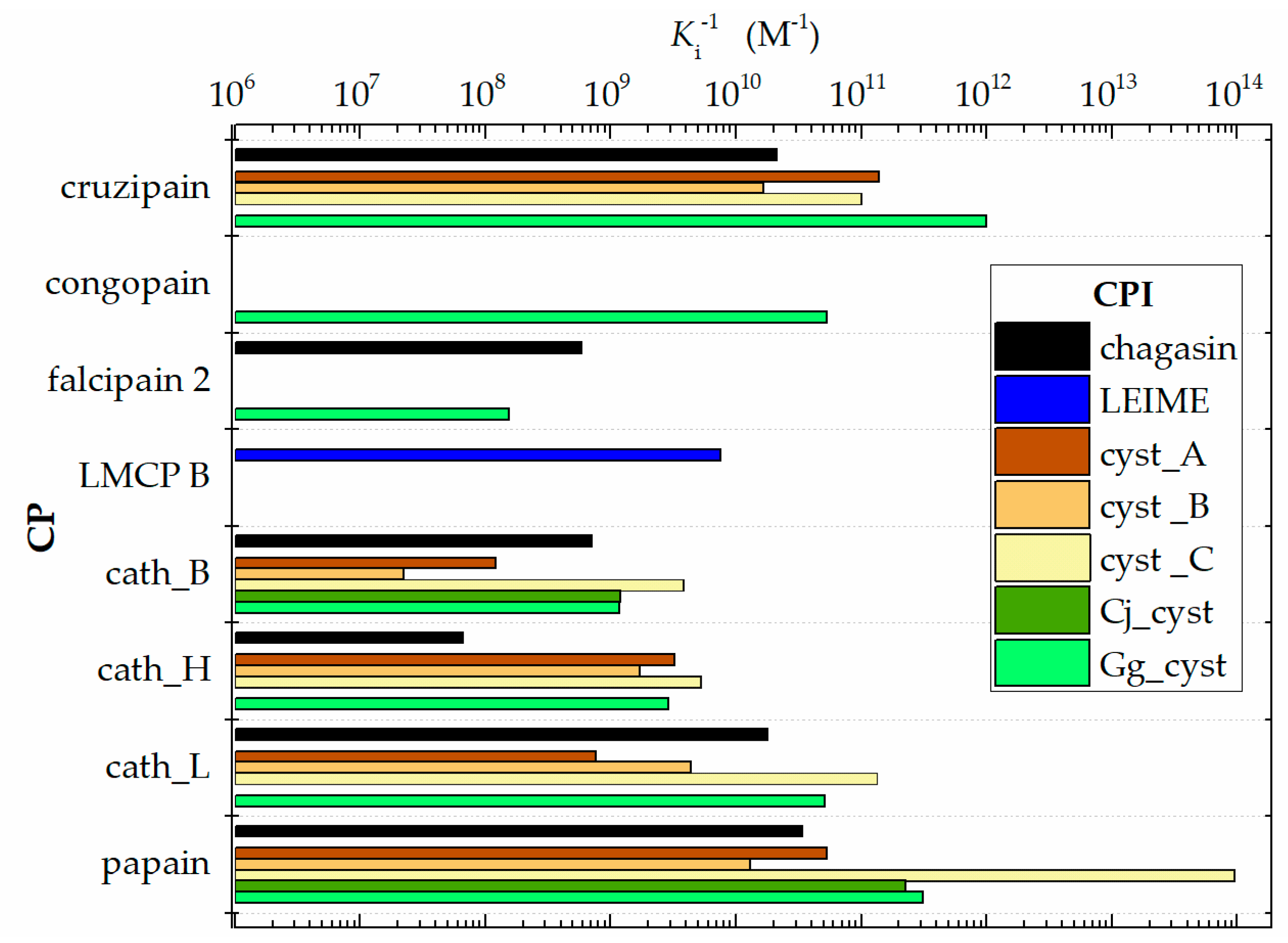




| Enzyme | Natural Inhibitor Cystein-Protease | ||||||
|---|---|---|---|---|---|---|---|
| Chagasin | LEIME | cyst_A | cyst _B | cyst _C | Cj_cyst | Gg_cyst | |
| cruzipain | 0.018 d,b | 0.007 a,k | 0.060 a | 0.01 a,h | 0.001 a,k | ||
| 0.076 i | 0.005 d | ||||||
| congopain | 0.019 f | ||||||
| falcipain 2 | 1.70 b,c | 6.50 c | |||||
| LMCP B | 0.133 j | ||||||
| cath_B | 0.93 b | 8.20 a | 73.00 a | 0.27 a,d | 0.828 g | 0.81 e | |
| 1.90 d | 16.00 b | 0.26 e | 0.047 g | ||||
| cath_H | 15.00 c | 0.31 a | 0.58 a | 0.28 a | 0.06 a | ||
| 0.10 e | 0.63 c | ||||||
| cath_L | 0.039 d,b | 1.30 a | 0.23 a | <0.005 a,d | 0.019 a,e | ||
| papain | 0.036 b | 0.019 a | 0.12 a | 0.00001 a,d | 0.004 g | 0.005 a,e | |
| 0.023 d | 0.03 b | 0.0014 g | |||||
| Model | E1_I1 | E2_I1 | E3_I1 | E4_I1 | E5_I1 | E6_I1 | E7_I1 | E8_I1 | E9_I1 | E10_I1 |
| Template a | 2NQD | 2NQD | 2NQD | 2NQD | 2NQD | 3CBK | 2NQD | 2NQD | 2NQD | 3E1Z |
| GMQE b | 0.84 | 0.83 | 0.78 | 0.84 | 0.84 | 0.99 | 0.84 | 0.98 | 0.97 | 0.99 |
| PROCOS c | 0.91 | 0.97 | 0.97 | 1.00 | 0.97 | 0.78 | 0.97 | 0.99 | 1.00 | 0.69 |
| Model | E1_I2 | E2_I2 | E3_I2 | E4_I2 | E5_I2 | E6_I2 | E7_I2 | E8_I2 | E9_I2 | E10_I2 |
| Template a | 2NQD | 2NQD | 3PNR | 2NQD | 2NQD | 3CBK | 2NQD | 2NQD | 2NQD | 3E1Z |
| GMQE b | 0.73 | 0.73 | 0.84 | 0.73 | 0.73 | 0.89 | 0.73 | 0.87 | 0.86 | 0.87 |
| PROCOS c | 0.97 | 0.97 | 0.61 | 0.99 | 0.97 | 0.62 | 0.99 | 0.97 | 1.0 | 0.97 |
| Model | E1_I3 | E2_I3 | E3_I3 | E4_I3 | E5_I3 | E6_I3 | E7_I3 | E8_I3 | E9_I3 | E10_I3 |
| Template a | 3KSE | 3KSE | 3K9M | 3KSE | 3KSE | 3K9M | 1NB5 | 3KSE | 3KSE | 3K9M |
| GMQE b | 0.83 | 0.83 | 0.71 | 0.83 | 0.83 | 0.99 | 0.98 | 0.98 | 0.97 | 0.75 |
| PROCOS c | 0.59 | 0.86 | 0.97 | 0.70 | 0.82 | 0.97 | 0.97 | 0.63 | 0.59 | 0.67 |
| Model | E1_I4 | E2_I4 | E3_I4 | E4_I4 | E5_I4 | E6_I4 | E7_I4 | E8_I4 | E9_I4 | E10_I4 |
| Template a | 3KSE | 3KSE | 3K9M | 3KSE | 3KSE | 3K9M | 1NB5 | 3KSE | 3KSE | 3K9M |
| GMQE b | 0.77 | 0.77 | 0.66 | 0.76 | 0.77 | 0.93 | 0.92 | 0.92 | 0.91 | 0.72 |
| PROCOS c | 0.86 | 0.97 | 0.97 | 0.91 | 0.98 | 0.78 | 0.97 | 0.64 | 0.82 | 0.82 |
| Model | E1_I5 | E2_I5 | E3_I5 | E4_I5 | E5_I5 | E6_I5 | E7_I5 | E8_I5 | E9_I5 | E10_I5 |
| Template a | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB |
| GMQE b | 0.74 | 0.75 | 0.92 | 0.73 | 0.74 | 0.64 | 0.74 | 0.74 | 0.74 | 0.74 |
| PROCOS c | 0.94 | 0.97 | 0.86 | 0.67 | 0.69 | 0.91 | 0.97 | 0.91 | 0.69 | 0.67 |
| Model | E1_I6 | E2_I6 | E3_I6 | E4_I6 | E5_I6 | E6_I6 | E7_I6 | E8_I6 | E9_I6 | E10_I6 |
| Template a | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB |
| GMQE b | 0.82 | 0.82 | 0.98 | 0.80 | 0.81 | 0.71 | 0.82 | 0.82 | 0.82 | 0.82 |
| PROCOS c | 0.97 | 0.97 | 1.00 | 0.97 | 0.97 | 0.97 | 0.97 | 0.97 | 0.99 | 0.82 |
| Model | E1_I7 | E2_I7 | E3_I7 | E4_I7 | E5_I7 | E6_I7 | E7_I7 | E8_I7 | E9_I7 | E10_I7 |
| Template a | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB | 1YVB |
| GMQE b | 0.81 | 0.81 | 0.99 | 0.81 | 0.81 | 0.82 | 0.82 | 0.71 | 0.82 | 0.82 |
| PROCOS c | 0.97 | 0.97 | 0.98 | 0.97 | 0.97 | 0.91 | 0.99 | 0.97 | 0.99 | 0.94 |
| PDB ID a | SM_HM | RMSD (Å) | Sequence Identity (%) | Scores PROCOS | |
|---|---|---|---|---|---|
| PDB | SM HM | ||||
| 2OUL | falcipain−chagasin | 1.22 | 100.0 | 0.97 | 0.97 |
| 1YVB | falcipain−Gg_cyst | 0.42 | 100.0 | 0.98 | 0.99 |
| 3CBK | cath_B−chagasin | 0.42 | 99.2 | 0.39 | 0.78 |
| 3K9M | cath_B−cyst_A | 0.45 | 100.0 | 0.97 | 0.97 |
| 2NQD | cath_L−chagasin | 0.38 | 99.6 | 0.97 | 0.99 |
| 3KSE | cath_L−cyst_A | 0.34 | 99.4 | 0.63 | 0.63 |
| 3KFQ | cath_V−cyst_A | 0.89 | 99.7 | 0.63 | 0.59 |
| 3E1Z | papain−chagasin | 0.35 | 100.0 | 0.82 | 0.69 |
| 1STF | papain−cyst_B | 1.20 | 96.5 | 0.97 | 0.82 |
| EC | Protease | Organism | ID | PDB a | Inhibitor | Organism | ID |
|---|---|---|---|---|---|---|---|
| 3.4.22.51 | cruzipain | T. cruzi | P25779 | 1ME3 b | |||
| congopain | T. congolense | Q26909 | |||||
| 3.4.22.B69 | falcipain 2 | P. falciparum | Q9N6S8 | 1YVB c | Gg_cyst | G. gallus | P01038 |
| falcipain 2 | P. falciparum | Q8I6U4 | 2OUL c | chagasin | T. cruzi | Q966X9 | |
| LMCP | L. mexicana | Q7JMY2 | |||||
| 3.4.22.B5 | LMCP B | L. mexicana | P36400 | ||||
| 3.4.22.1 | cath_B | H. sapiens | P07858 | 3CBK c | chagasin | T. cruzi | Q966X9 |
| 3.4.22.1 | cath_B | H. sapiens | P07858 | 3K9M c | cyst_A | H. sapiens | P01040 |
| 3.4.22.16 | cath_H | H. sapiens | P09668 | ||||
| 3.4.22.15 | cath_L | H. sapiens | P07711 | 2NQD c | chagasin | T. cruzi | Q966X9 |
| 3.4.22.15 | cath_L | H. sapiens | P07711 | 3KSE c | cyst_A | H. sapiens | P01040 |
| 3.4.22.43 | cath_V | H. sapiens | O60911 | 3KFQ c | cyst_A | H. sapiens | P01040 |
| 3.4.22.2 | papain | C. papaya | P00784 | 3E1Z c | chagasin | T. cruzi | Q966X9 |
| 3.4.22.2 | papain | C. papaya | P00784 | 1STF c | cyst_B | H. sapiens | P04080 |
| 3.4.22.6 | chymo | C. papaya | P14080 | 1YAL b | |||
| 3.4.22.30 | caricain | C. papaya | P10056 | 1PPO b | |||
| Cj_cyst | C. japonica | P81061 | |||||
| 3GAX b | cyst_C | H. sapiens | P01034 | ||||
| 2C34 b | LEIME | L. mexicana | Q868H1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes-Espinosa, F.; Juárez-Saldivar, A.; Palos, I.; Herrera-Mayorga, V.; García-Pérez, C.; Rivera, G. In Silico Analysis of Homologous Heterodimers of Cruzipain-Chagasin from Structural Models Built by Homology. Int. J. Mol. Sci. 2019, 20, 1320. https://doi.org/10.3390/ijms20061320
Reyes-Espinosa F, Juárez-Saldivar A, Palos I, Herrera-Mayorga V, García-Pérez C, Rivera G. In Silico Analysis of Homologous Heterodimers of Cruzipain-Chagasin from Structural Models Built by Homology. International Journal of Molecular Sciences. 2019; 20(6):1320. https://doi.org/10.3390/ijms20061320
Chicago/Turabian StyleReyes-Espinosa, Francisco, Alfredo Juárez-Saldivar, Isidro Palos, Verónica Herrera-Mayorga, Carlos García-Pérez, and Gildardo Rivera. 2019. "In Silico Analysis of Homologous Heterodimers of Cruzipain-Chagasin from Structural Models Built by Homology" International Journal of Molecular Sciences 20, no. 6: 1320. https://doi.org/10.3390/ijms20061320