Association of Human FOS Promoter Variants with the Occurrence of Knee-Osteoarthritis in a Case Control Association Study

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Initial SNP Analysis in Rheumatoid Arthritis, Knee-Osteoarthritis, and NC Samples
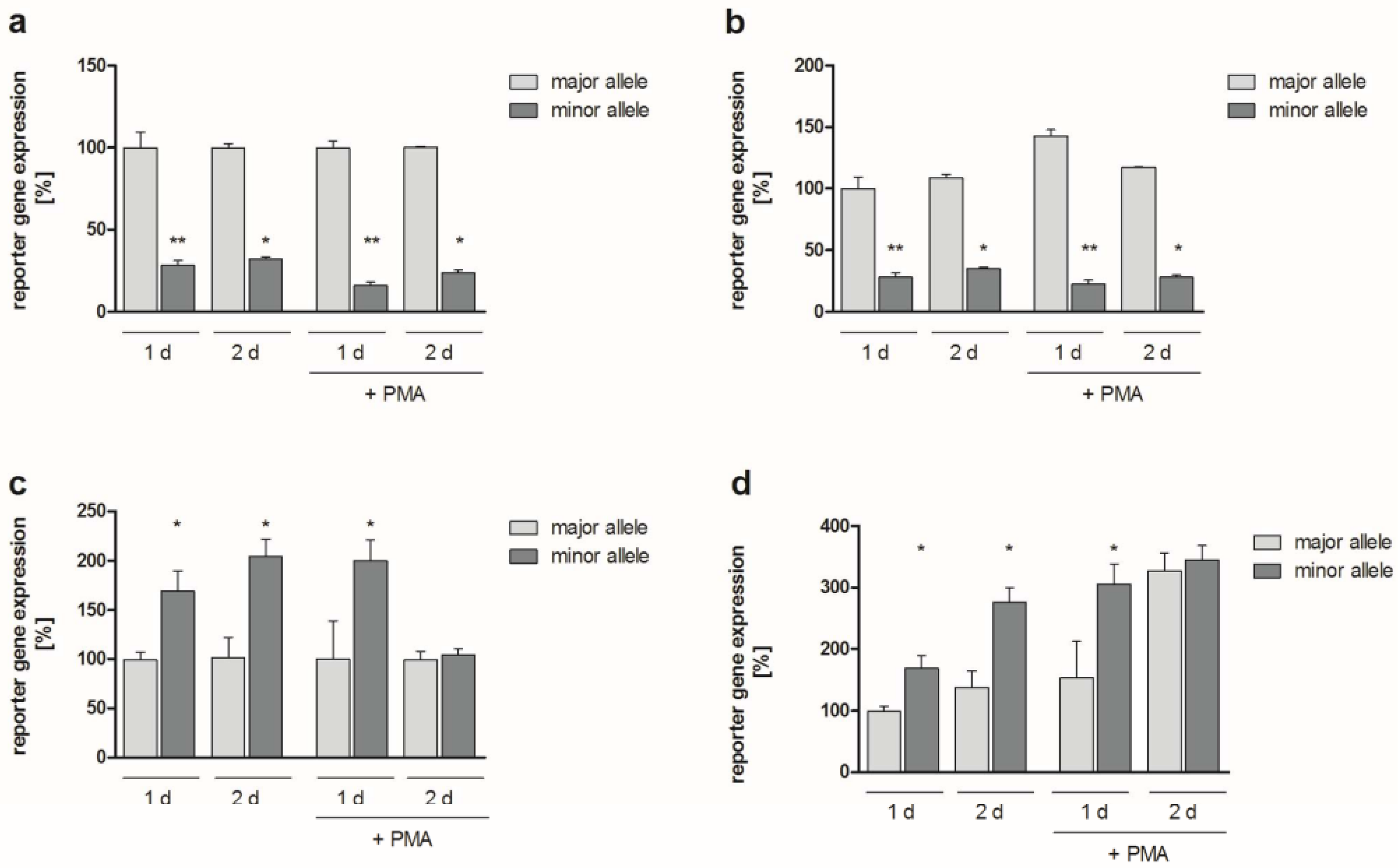
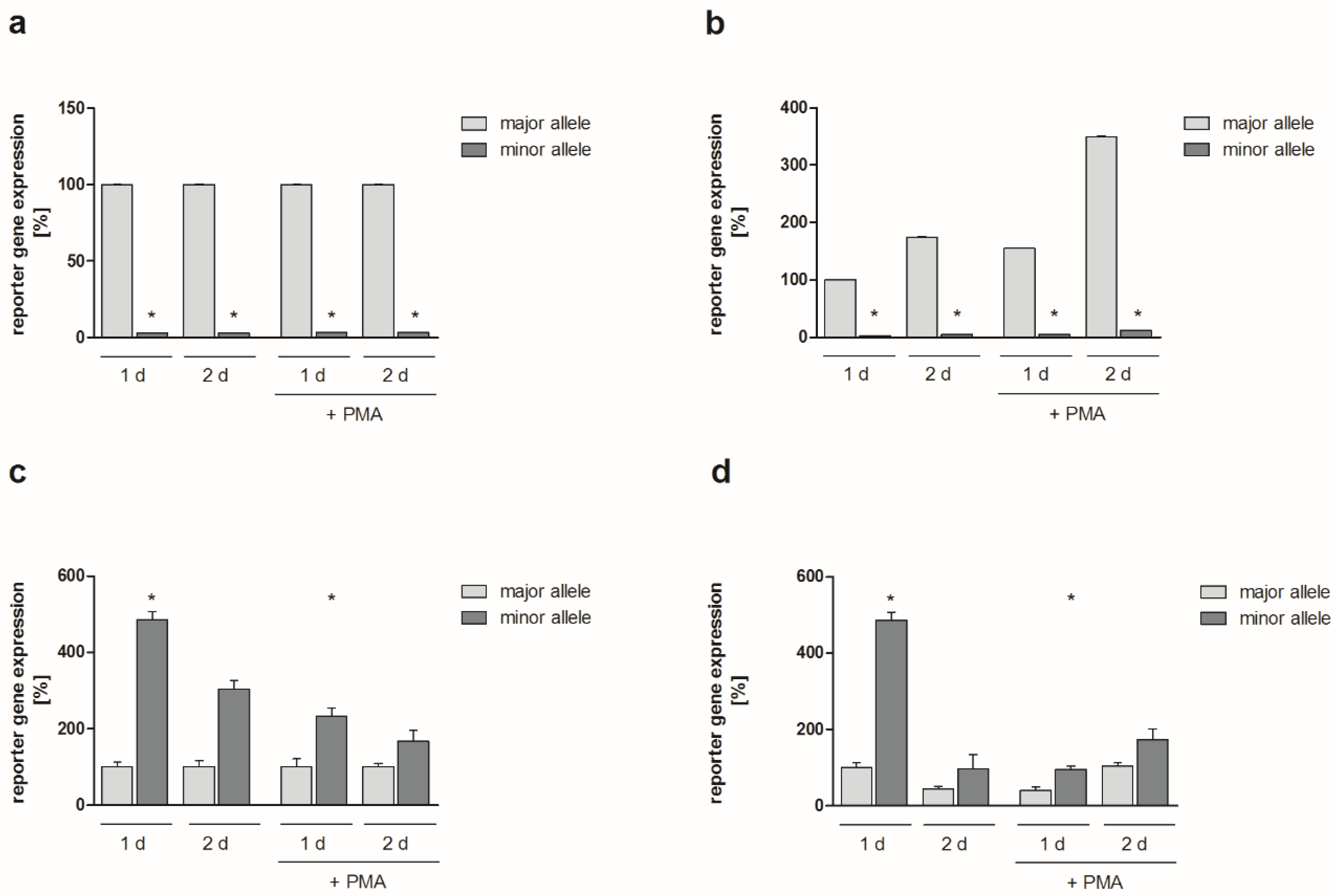
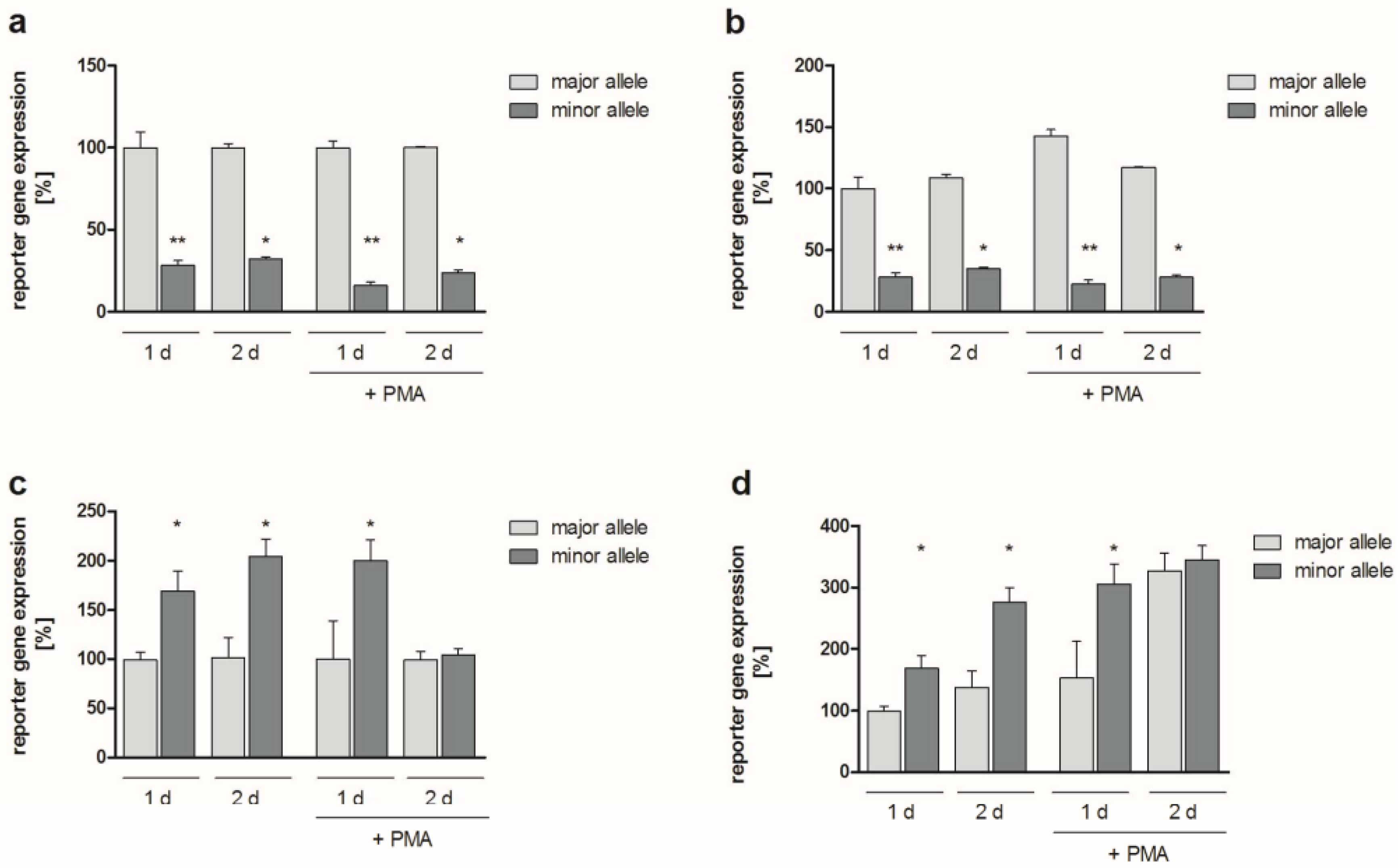
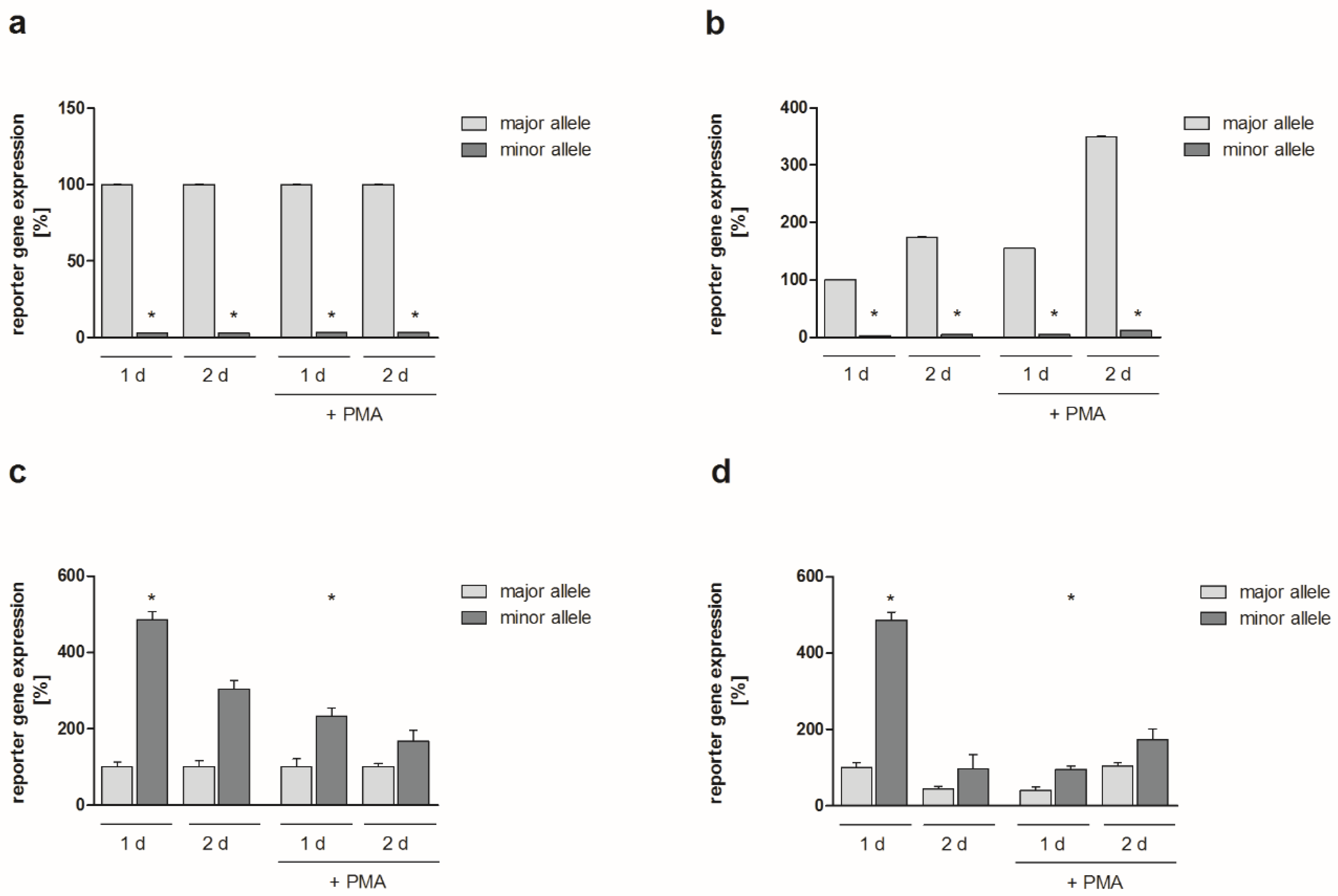
2.2. Functional Analyses
2.3. Allelic Distribution of Polymorphisms and Statistical Analysis within the German Cohort
2.4. Replication of the Genetic Association within the Finnish Cohort
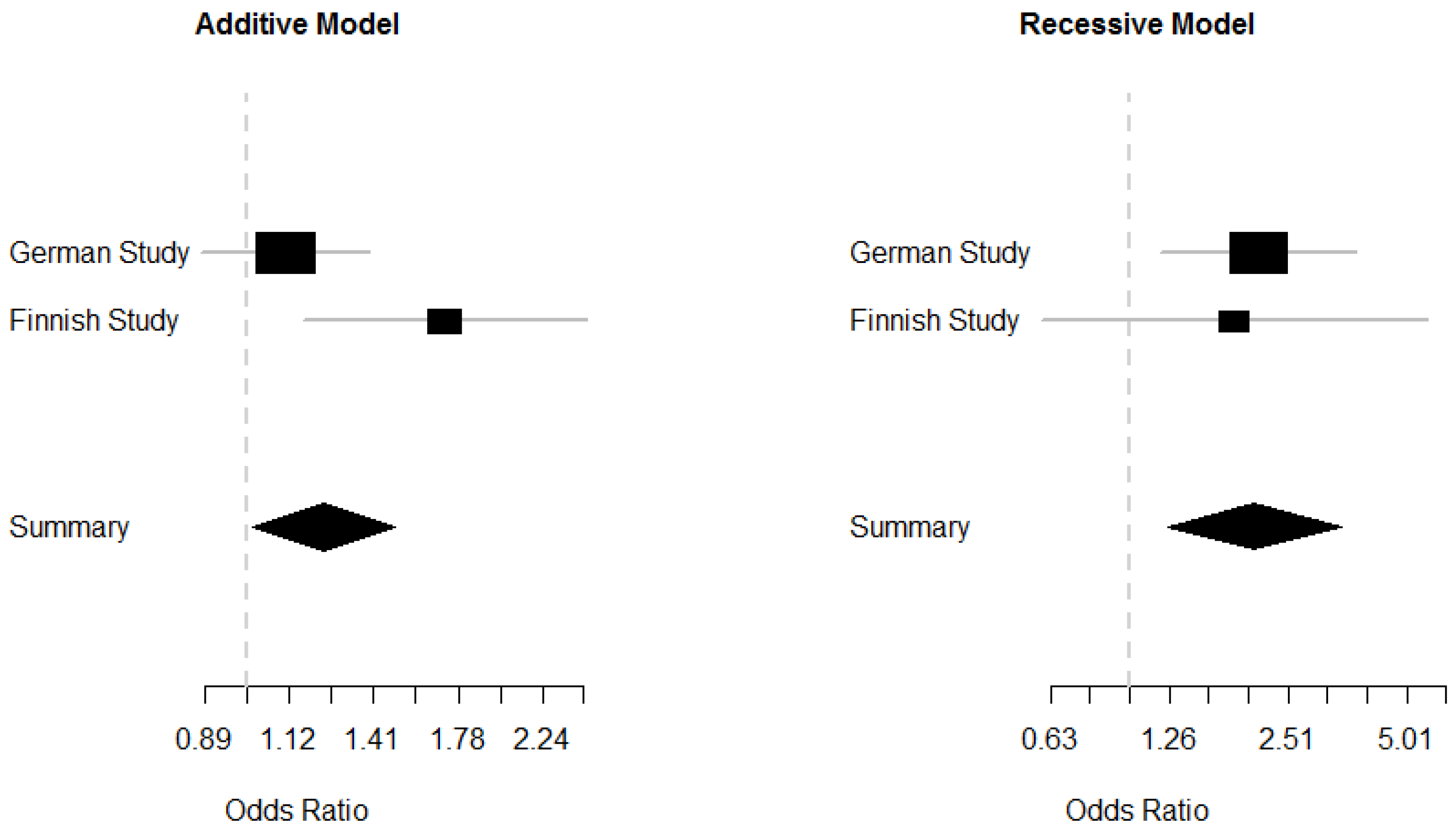
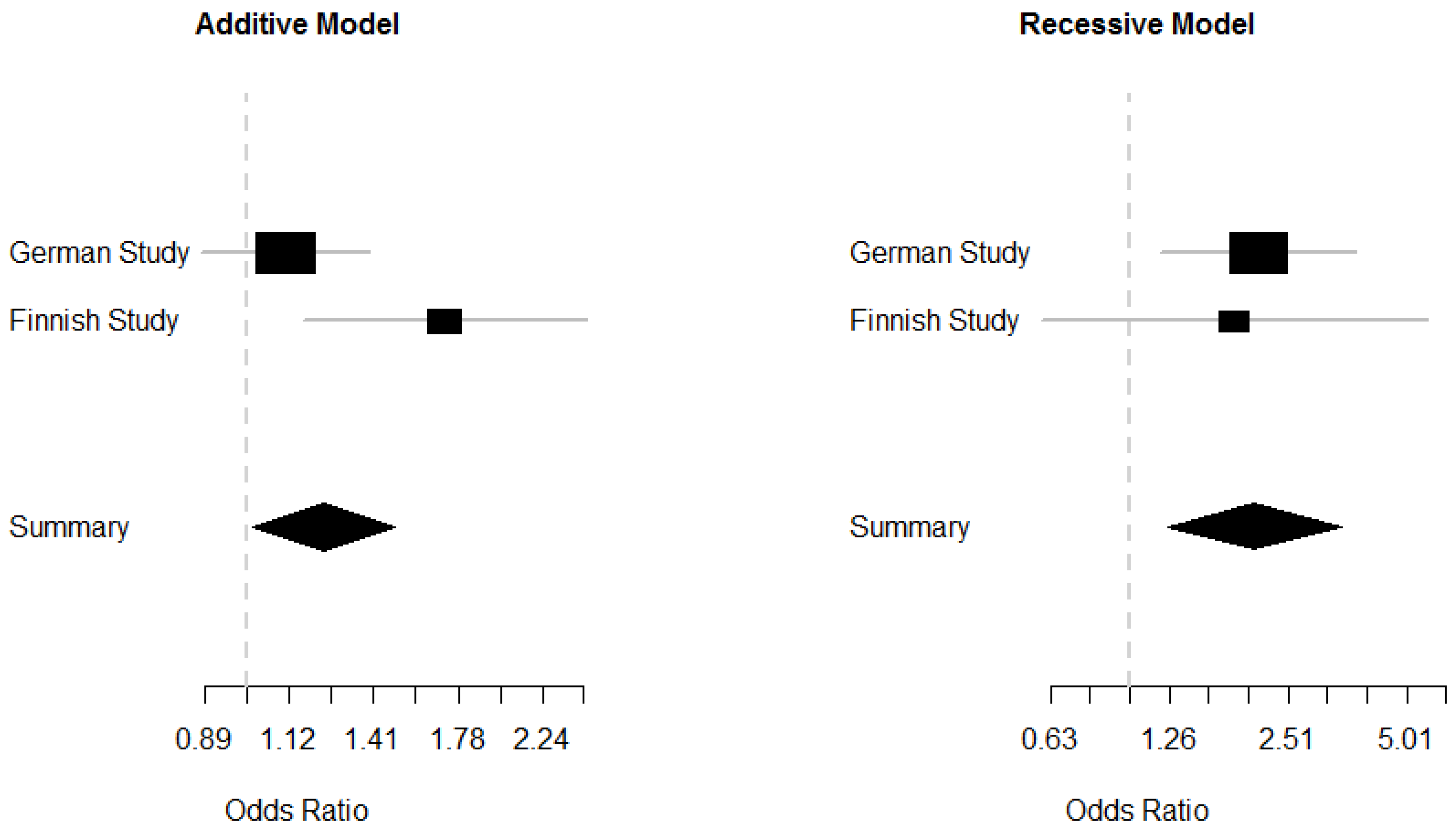
2.5. Meta-Analysis of the German and Finnish Studies
3. Discussion
4. Materials and Methods
4.1. Patients and Tissue Samples
4.2. DNA Preparation
4.3. Conventional PCR
4.4. Cloning of DNA Fragments
4.5. Non-Isotopic RNase Cleavage Assay
4.6. Functional Analyses
4.7. Genotyping and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AP-1 | activator protein 1 |
| CI | confidence interval |
| EP300 | 300 kDa E1A-binding protein |
| FOS | v-Fos FBJ murine osteosarcoma viral oncogene homolog |
| GWAS | genome-wide association studies |
| HEY1 | hairy/enhancer of split-related with YRPW motif 1 |
| IL1B | interleukin 1β |
| JUN | v-Jun avian sarcoma virus 17 oncogene homolog |
| JUNB | oncogene Jun-B |
| JUND | oncogene Jun-D |
| LD | linkage disequilibrium |
| MAF | minor allele frequency |
| MALDI-TOF | matrix-assisted laser desorption/ionization—time of flight |
| NC | normal controls |
| NIRCA | non-isotopic RNase cleavage assay |
| OA | osteoarthritis |
| OR | odds ratio |
| PMA | phorbol-12-myristate 13-acetate |
| POU2F2 | POU domain class 2 transcription factor 2 |
| RA | rheumatoid arthritis |
| RLA | relative luciferase activity |
| RLU | relative light units |
| SEM | standard error of the mean |
| SM | synovial membrane |
| SNP | single nucleotide polymorphism |
| SRF | serum response factor |
| TAF1 | TATA box binding protein-associated factor 1 |
References
- Kinne, R.W.; Palombo-Kinne, E.; Emmrich, F. Activation of synovial fibroblasts in rheumatoid arthritis. Ann. Rheum. Dis. 1995, 54, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, J.C.; Delmas, P.D. Biological markers in osteoarthritis. Nat. Clin. Pract. Rheumatol. 2007, 3, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.P. Osteoarthritis. Nat. Rev. Dis. Primers 2016, 2, 16072. [Google Scholar] [CrossRef] [Green Version]
- Lories, R.J. Joint homeostasis, restoration, and remodeling in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2008, 22, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; Manning, A.M. Signal transduction and transcription factors in rheumatic disease. Arthritis Rheum. 1999, 42, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Angel, P.; Karin, M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [Google Scholar] [CrossRef]
- Rengel, Y.; Ospelt, C.; Gay, S. Proteinases in the joint: Clinical relevance of proteinases in joint destruction. Arthritis Res. Ther. 2007, 9, 221. [Google Scholar] [CrossRef]
- Burrage, P.S.; Brinckerhoff, C.E. Molecular targets in osteoarthritis: Metalloproteinases and their inhibitors. Curr. Drug Targets 2007, 8, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, S.; Tsumiyama, K. Pathogenesis of rheumatoid arthritis and c-Fos/AP-1. Cell Cycle 2009, 8, 1539–1543. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, M.; Hirakawa, K.; Kato, A.; Fujii, K. The possible role of c-fos expression in rheumatoid cartilage destruction. J. Rheumatol. 2000, 27, 1606–1621. [Google Scholar]
- Shaulian, E.; Karin, M. AP-1 in cell proliferation and survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef] [Green Version]
- Ransone, L.J.; Verma, I.M. Nuclear proto-oncogenes fos and jun. Annu. Rev. Cell Biol. 1990, 6, 539–557. [Google Scholar] [CrossRef]
- Bax, M.; van Heemst, J.; Huizinga, T.W.; Toes, R.E. Genetics of rheumatoid arthritis: What have we learned? Immunogenetics 2011, 63, 459–466. [Google Scholar] [CrossRef]
- Chapman, K.; Valdes, A.M. Genetic factors in OA pathogenesis. Bone 2011, 51, 258–264. [Google Scholar] [CrossRef]
- Harrison, P.; Pointon, J.J.; Chapman, K.; Roddam, A.; Wordsworth, B.P. Interleukin-1 promoter region polymorphism role in rheumatoid arthritis: A meta-analysis of IL-1B-511A/G variant reveals association with rheumatoid arthritis. Rheumatology 2008, 47, 1768–1770. [Google Scholar] [CrossRef]
- Rueda, B.; Oliver, J.; Robledo, G.; Lopez-Nevot, M.A.; Balsa, A.; Pascual-Salcedo, D.; Gonzalez-Gay, M.A.; Gonzalez-Escribano, M.F.; Martin, J. HO-1 promoter polymorphism associated with rheumatoid arthritis. Arthritis Rheum. 2007, 56, 3953–3958. [Google Scholar] [CrossRef]
- Southam, L.; Rodriguez-Lopez, J.; Wilkins, J.M.; Pombo-Suarez, M.; Snelling, S.; Gomez-Reino, J.J.; Chapman, K.; Gonzalez, A.; Loughlin, J. An SNP in the 5′-UTR of GDF5 is associated with osteoarthritis susceptibility in Europeans and with in vivo differences in allelic expression in articular cartilage. Hum. Mol. Genet 2007, 16, 2226–2232. [Google Scholar] [CrossRef]
- Mahr, S.; Muller-Hilke, B. Transcriptional activity of the RHOB gene is influenced by regulatory polymorphisms in its promoter region. Genom. Med. 2007, 1, 125–128. [Google Scholar] [CrossRef] [Green Version]
- dbSNP build 130. Available online: http://www.ncbi.nlm.nih.gov/projects/SNP (accessed on 1 February 2010).
- Asahara, H.; Fujisawa, K.; Kobata, T.; Hasunuma, T.; Maeda, T.; Asanuma, M.; Ogawa, N.; Inoue, H.; Sumida, T.; Nishioka, K. Direct evidence of high DNA binding activity of transcription factor AP-1 in rheumatoid arthritis synovium. Arthritis Rheum. 1997, 40, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 1998, 28, 197–208. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Firestein, G.S. Pathogenesis of rheumatoid arthritis: The role of synoviocytes. Rheum. Dis. Clin. N. Am. 2001, 27, 355–371. [Google Scholar] [CrossRef]
- Chiu, Y.C.; Yang, R.S.; Hsieh, K.H.; Fong, Y.C.; Way, T.D.; Lee, T.S.; Wu, H.C.; Fu, W.M.; Tang, C.H. Stromal cell-derived factor-1 induces matrix metalloprotease-13 expression in human chondrocytes. Mol. Pharmacol. 2007, 72, 695–703. [Google Scholar] [CrossRef]
- Benderdour, M.; Tardif, G.; Pelletier, J.P.; di Battista, J.A.; Reboul, P.; Ranger, P.; Martel-Pelletier, J. Interleukin 17 (IL-17) induces collagenase-3 production in human osteoarthritic chondrocytes via AP-1 dependent activation: differential activation of AP-1 members by IL-17 and IL-1beta. J. Rheumatol. 2002, 29, 1262–1272. [Google Scholar]
- Zafarullah, M.; Martel-Pelletier, J.; Cloutier, J.M.; Gedamu, L.; Pelletier, J.P. Expression of c-fos, c-jun, jun-B, metallothionein and metalloproteinase genes in human chondrocyte. FEBS Lett. 1992, 306, 169–172. [Google Scholar] [CrossRef]
- Zafarullah, M.; Martel-Pelletier, J.; Cloutier, J.M.; Gedamu, L.; Pelletier, J.P. Distinct expression pattern of early- and late-response genes in normal and osteoarthritic human synovial membranes. Osteoarthr. Cartil. 1993, 1, 151–156. [Google Scholar] [CrossRef]
- Celniker, S.E.; Dillon, L.A.; Gerstein, M.B.; Gunsalus, K.C.; Henikoff, S.; Karpen, G.H.; Kellis, M.; Lai, E.C.; Lieb, J.D.; MacAlpine, D.M.; et al. Unlocking the secrets of the genome. Nature 2009, 459, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Myers, A.J.; Gibbs, J.R.; Webster, J.A.; Rohrer, K.; Zhao, A.; Marlowe, L.; Kaleem, M.; Leung, D.; Bryden, L.; Nath, P.; et al. A survey of genetic human cortical gene expression. Nat. Genet. 2007, 39, 1494–1499. [Google Scholar] [CrossRef]
- Karouzakis, E.; Neidhart, M.; Gay, R.E.; Gay, S. Molecular and cellular basis of rheumatoid joint destruction. Immunol. Lett. 2006, 106, 8–13. [Google Scholar] [CrossRef]
- Manke, T.; Heinig, M.; Vingron, M. Quantifying the effect of sequence variation on regulatory interactions. Hum. Mutat. 2010, 31, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31, 315–324. [Google Scholar] [CrossRef]
- Altman, R.; Asch, E.; Bloch, D.; Bole, G.; Borenstein, D.; Brandt, K.; Christy, W.; Cooke, T.D.; Greenwald, R.; Hochberg, M. Development of criteria for the classification and reporting of osteoarthritis. Classification of osteoarthritis of the knee. Diagnostic and Therapeutic Criteria Committee of the American Rheumatism Association. Arthritis Rheum. 1986, 29, 1039–1049. [Google Scholar] [CrossRef]
- Perttila, J.; Merikanto, K.; Naukkarinen, J.; Surakka, I.; Martin, N.W.; Tanhuanpaa, K.; Grimard, V.; Taskinen, M.R.; Thiele, C.; Salomaa, V.; et al. OSBPL10, a novel candidate gene for high triglyceride trait in dyslipidemic Finnish subjects, regulates cellular lipid metabolism. J. Mol. Med. 2009, 87, 825–835. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.; Kunisch, E.; Gluck, B.; Egerer, R.; Sickinger, S.; Kinne, R.W. Comparison of conventional and real-time RT-PCR for the quantitation of jun protooncogene mRNA and analysis of junB mRNA expression in synovial membranes and isolated synovial fibroblasts from rheumatoid arthritis patients. Z. Rheumatol. 2003, 62, 378–389. [Google Scholar] [CrossRef]
- De Martin, R.; Strasswimmer, J.; Philipson, L. A new luciferase promoter insertion vector for the analysis of weak transcriptional activities. Gene 1993, 124, 137–138. [Google Scholar] [CrossRef]
- Dunger, S.; Neumann, S.; Zell, R.; Birch-Hirschfeld, E.; Stelzner, A.; Paschke, R.; Kinne, R.W.; Sickinger, S. Mutation detection in mosaic situations: RNA mismatch assay and denaturing gradient gel electrophoresis are more sensitive than conventional cycle sequencing. Anal. Biochem. 2001, 294, 89–93. [Google Scholar] [CrossRef]
- Poxon, S.W.; Hughes, J.A. A biofunctional assay to study pRL-CMV plasmid DNA formulation stability. PDA J. Pharm. Sci. Technol. 1999, 53, 314–317. [Google Scholar]
- Kirsten, H.; Dienst, S.; Emmrich, F.; Ahnert, P. CalcDalton: A tool for multiplex genotyping primer design for single-base extension reactions using cleavable primers. Biotechniques 2006, 40, 158–162. [Google Scholar] [CrossRef]
- Kirsten, H.; Teupser, D.; Weissfuss, J.; Wolfram, G.; Emmrich, F.; Ahnert, P. Robustness of single-base extension against mismatches at the site of primer attachment in a clinical assay. J. Mol. Med. 2007, 85, 361–369. [Google Scholar] [CrossRef]
- Lathrop, G.M. Estimating genotype relative risks. Tissue Antigens 1983, 22, 160–166. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Altman, D.G.; Bland, J.M. Interaction revisited: The difference between two estimates. BMJ 2003, 326, 219. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Variable | NC Cohort | RA Cohort | OA Cohort |
|---|---|---|---|
| rs4647009 | |||
| (CA/CA) | 0% (0) | 0% (0) | 0.4% (1) |
| (CA/−) | 11.0% (53) | 11.6% (34) | 9.7% (27) |
| (−/−) | 89.0% (427) | 88.4% (260) | 89.9% (249) |
| HWE p-value | 0.2 | 0.29 | 0.78 |
| Allelic OR (95% CI) | / | 1.05 (0.7–1.6) | 0.95 (0.6–1.5) |
| Allelic OR p-value | / | 0.84 | 0.82 |
| rs7101 | |||
| Homozygous Minor (C/C) | 5.0% (24) | 5.1% (15) | 10.1% (28) |
| Heterozygous (C/T) | 35.1% (167) | 40.3% (119) | 29.2% (81) |
| Homozygous Major (T/T) | 59.9% (285) | 54.6% (161) | 60.6% (168) |
| HWE p-value | 0.98 | 0.24 | 0.0006 |
| Allelic OR (95% CI) | / | 1.16 (0.9–1.5) | 1.13 (0.9–1.4) |
| Allelic OR p-value | / | 0.23 | 0.35 |
| Minor Recessive OR (95% CI) | / | 1.01 (0.5–2.0) | 2.12 (1.2–3.7) |
| Minor Recessive OR p-value | / | 0.99 | 0.0086 |
| Variable | NC Cohort | OA Cohort |
|---|---|---|
| Homozygous Minor (C/C) | 17 | 4 |
| Heterozygous (C/T) | 184 | 35 |
| Homozygous Major (T/T) | 347 | 33 |
| HWE p-value | 0.26 | 0.23 |
| allelic OR (95% CI) | / | 1.72 (1.2–2.5) |
| allelic OR p-value | / | 0.0058 |
| Minor Recessive OR (95% CI) | / | 1.84 (0.6–5.6) |
| Minor Recessive OR p-value | / | 0.29 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huber, R.; Kirsten, H.; Näkki, A.; Pohlers, D.; Thude, H.; Eidner, T.; Heinig, M.; Brand, K.; Ahnert, P.; Kinne, R.W. Association of Human FOS Promoter Variants with the Occurrence of Knee-Osteoarthritis in a Case Control Association Study. Int. J. Mol. Sci. 2019, 20, 1382. https://doi.org/10.3390/ijms20061382
Huber R, Kirsten H, Näkki A, Pohlers D, Thude H, Eidner T, Heinig M, Brand K, Ahnert P, Kinne RW. Association of Human FOS Promoter Variants with the Occurrence of Knee-Osteoarthritis in a Case Control Association Study. International Journal of Molecular Sciences. 2019; 20(6):1382. https://doi.org/10.3390/ijms20061382
Chicago/Turabian StyleHuber, René, Holger Kirsten, Annu Näkki, Dirk Pohlers, Hansjörg Thude, Thorsten Eidner, Matthias Heinig, Korbinian Brand, Peter Ahnert, and Raimund W. Kinne. 2019. "Association of Human FOS Promoter Variants with the Occurrence of Knee-Osteoarthritis in a Case Control Association Study" International Journal of Molecular Sciences 20, no. 6: 1382. https://doi.org/10.3390/ijms20061382
APA StyleHuber, R., Kirsten, H., Näkki, A., Pohlers, D., Thude, H., Eidner, T., Heinig, M., Brand, K., Ahnert, P., & Kinne, R. W. (2019). Association of Human FOS Promoter Variants with the Occurrence of Knee-Osteoarthritis in a Case Control Association Study. International Journal of Molecular Sciences, 20(6), 1382. https://doi.org/10.3390/ijms20061382