Interactions between Membrane Resistance, GABA-A Receptor Properties, Bicarbonate Dynamics and Cl−-Transport Shape Activity-Dependent Changes of Intracellular Cl− Concentration


Abstract
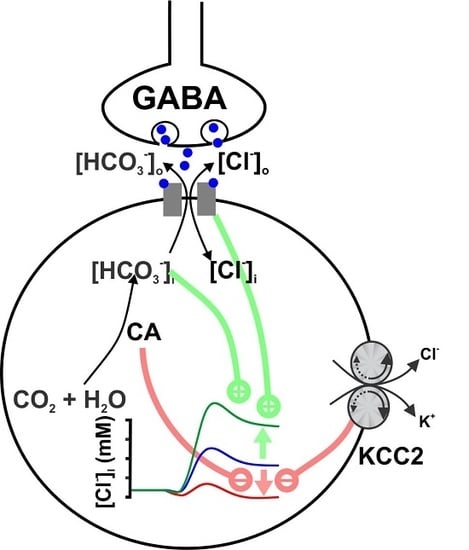
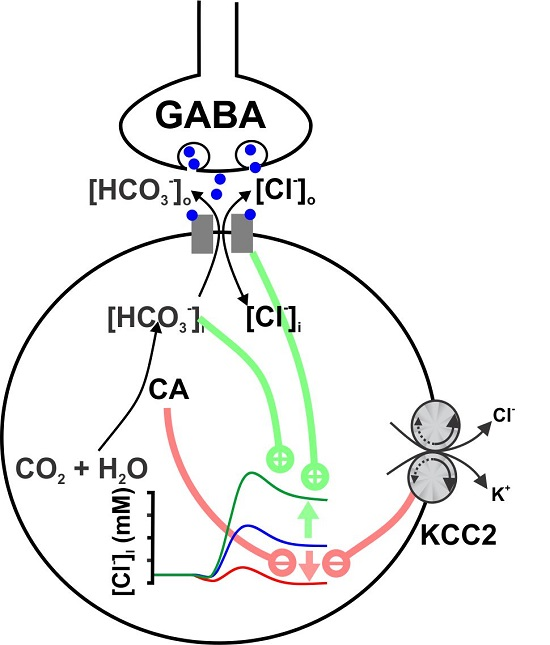
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
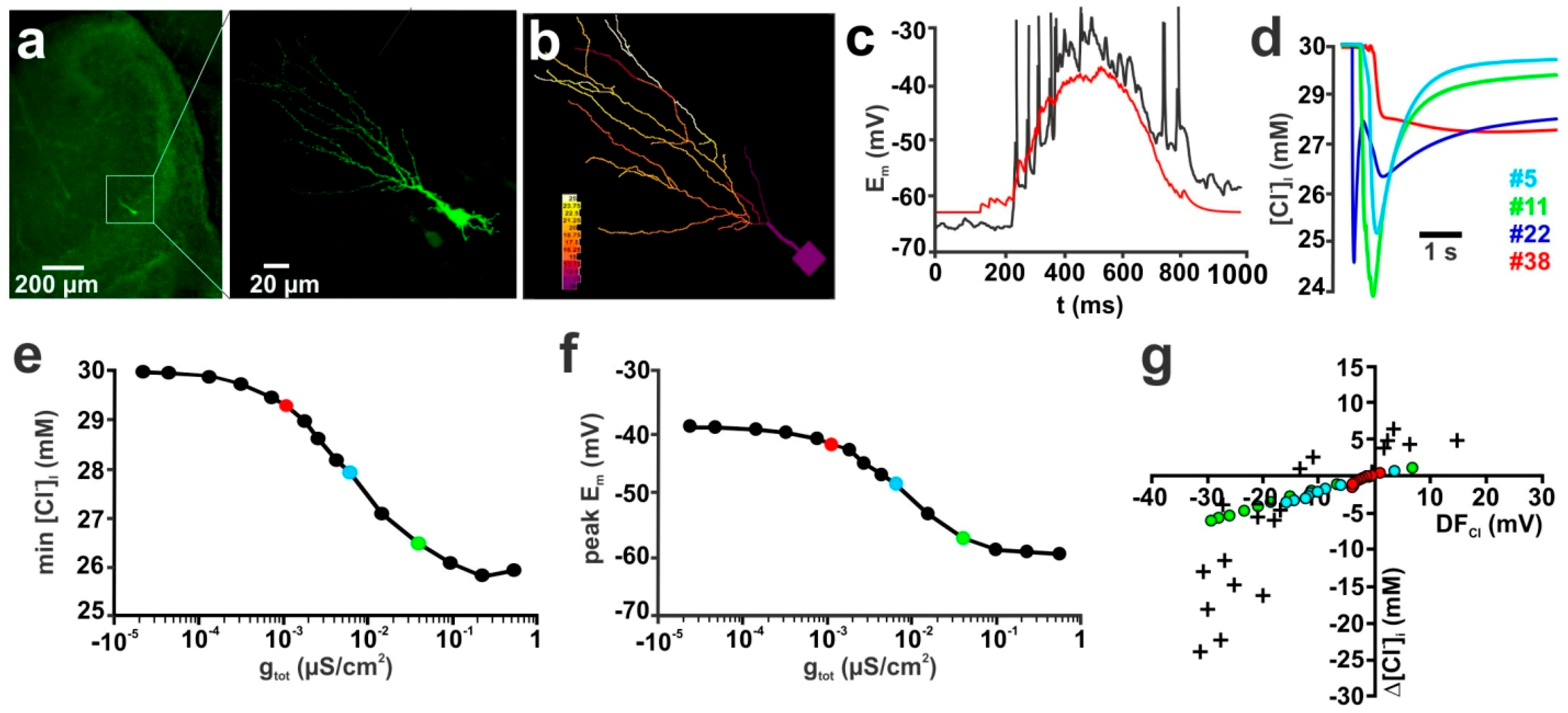
2.1. Influence of Membrane Conductance
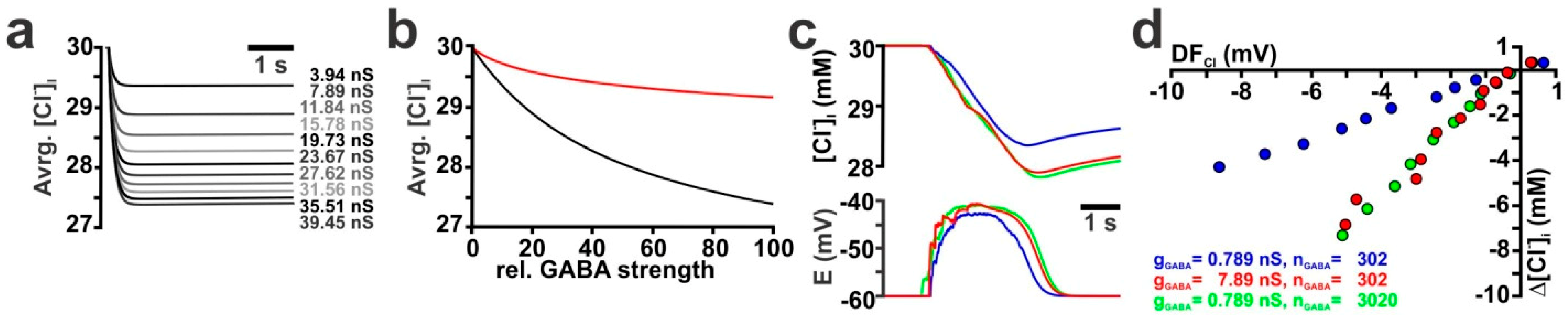
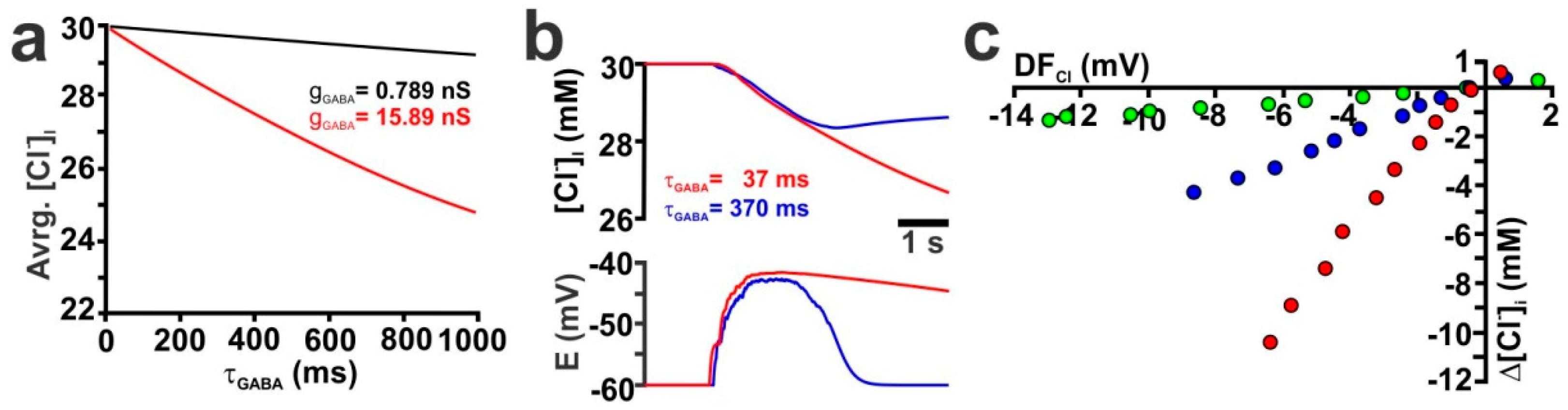
2.2. Influence of GABA Receptor Conductivity and Kinetics
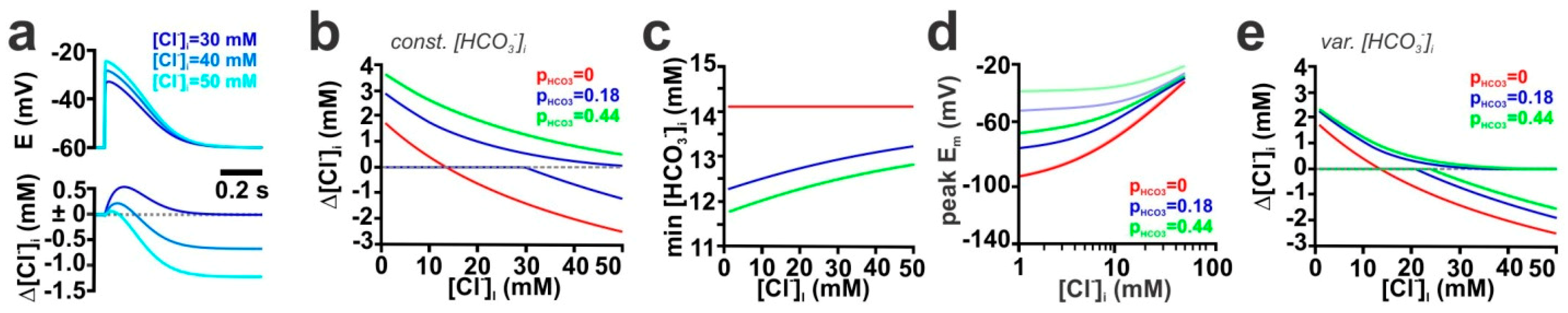
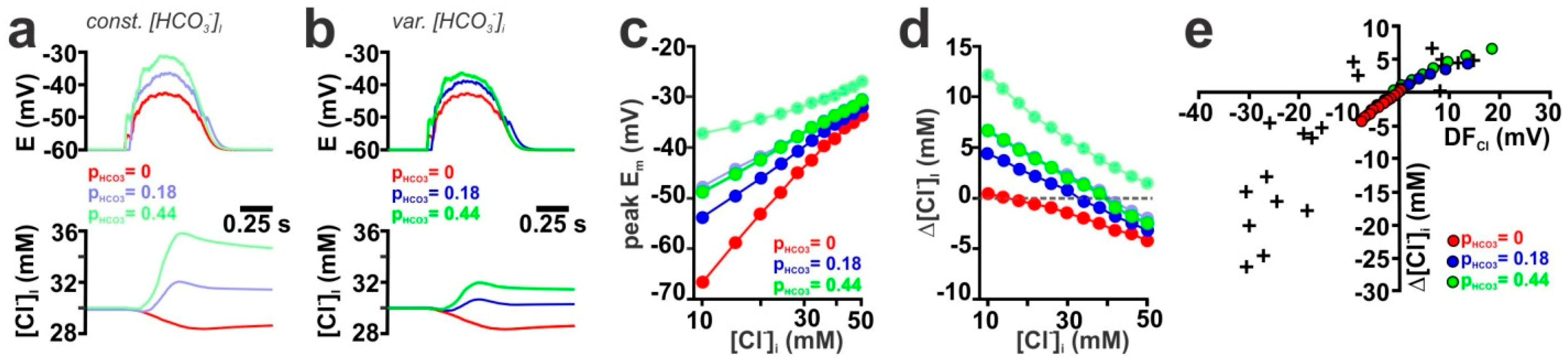
2.3. Contribution of the HCO3− Conductance of GABA Receptors
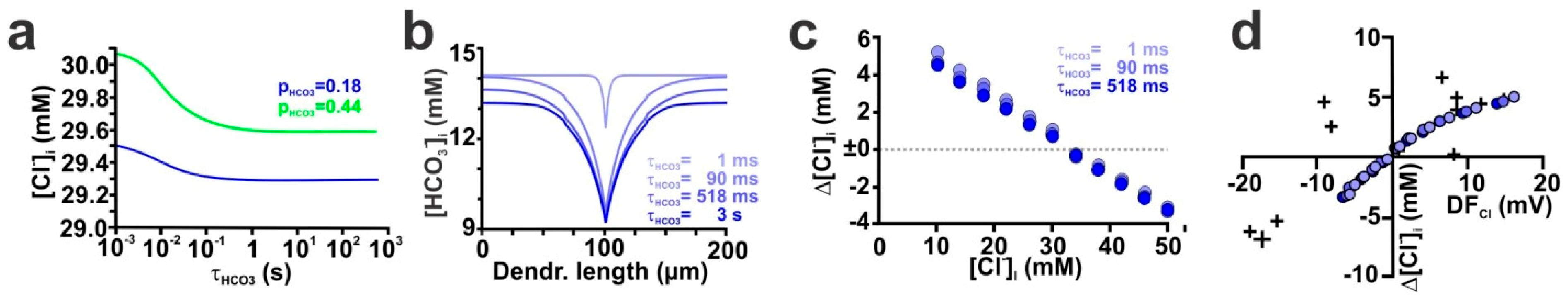
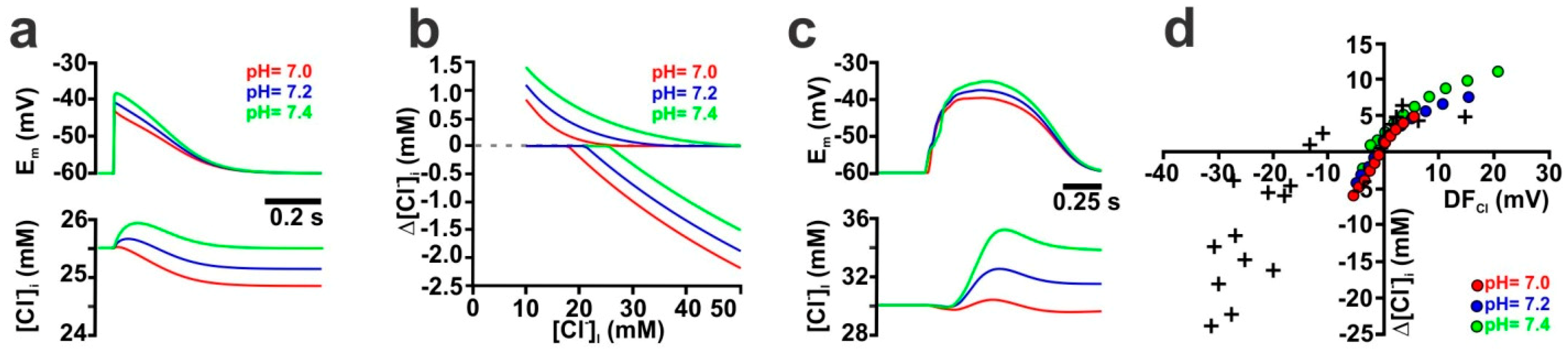
2.4. The Stability of HCO3− Gradients Influences Activity-Dependent [Cl−]i Transients
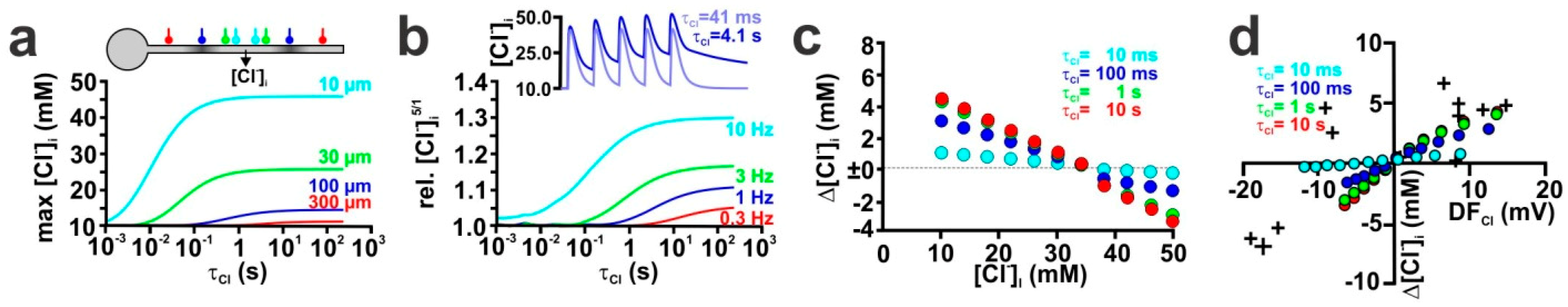
2.5. Influence of Transmembrane Cl− Transport
3. Discussion
4. Materials and Methods
Compartmental Modeling
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DFCl | Electromotive driving force on Cl− ions |
| ECl | Equilibrium potential for Cl− |
| EGABA | Reversal potential of GABAergic currents |
| EHCO3 | Equilibrium potential for HCO3− |
| GABA | γ-Amino butyric acid |
| GDP | Giant depolarizing potential |
| gGABA | Conductance of GABAergic synapse |
| gpas | Passive membrane conductance |
| KCC | K+-Cl−-Cotransporter |
| NKCC1 | Na+-K+-Cl−-Cotransporter, Isoform 1 |
| nGABA | Number of GABAergic synapses |
| PHCO3 | Relative HCO3− permeability of GABAA receptors |
| VHold | Holding potential |
| τCl | Time constant of [Cl−] relaxation |
| τGABA | Decay time constant of GABAA receptors |
| τHCO3 | Time constant of [HCO3−] relaxation |
References
- Farrant, M.; Kaila, K. The cellular, molecular and ionic basis of GABA(A) receptor signaling. Prog. Brain Res. 2007, 160, 59–87. [Google Scholar] [PubMed]
- Rivera, C.; Voipio, J.; Payne, J.A.; Ruusuvuori, E.; Lahtinen, H.; Lamsa, K.; Pirvola, U.; Saarma, M.; Kaila, K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 1999, 397, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Blaesse, P.; Airaksinen, M.S.; Rivera, C.; Kaila, K. Cation-chloride cotransporters and neuronal function. Neuron 2009, 61, 820–838. [Google Scholar] [CrossRef]
- Kaila, K.; Pasternack, M.; Saarikoski, J.; Voipio, J. Influence of GABA-gated bicarbonate conductance on potential, current and intracellular chloride in crayfish muscle fibres. J. Physiol. 1989, 416, 161–181. [Google Scholar] [CrossRef] [PubMed]
- Bracci, E.; Vreugdenhil, M.; Hack, S.P.; Jefferys, J.G.R. Dynamic modulation of excitation and inhibition during stimulation at gamma and beta frequencies in the CA1 hippocampal region. J. Neurophysiol. 2001, 85, 2412–2422. [Google Scholar] [CrossRef]
- Isomura, Y.; Sugimoto, M.; Fujiwara-Tsukamoto, Y.; Yamamoto-Muraki, S.; Yamada, J.; Fukuda, A. Synaptically activated Cl− accumulation responsible for depolarizing GABAergic responses in mature hippocampal neurons. J. Neurophysiol. 2003, 90, 2752–2756. [Google Scholar] [CrossRef]
- Jedlicka, P.; Deller, T.; Gutkin, B.S.; Backus, K.H. Activity-Dependent Intracellular Chloride Accumulation and Diffusion Controls GABA(A) Receptor-Mediated Synaptic Transmission. Hippocampus 2011, 21, 885–898. [Google Scholar]
- Lillis, K.P.; Kramer, M.A.; Mertz, J.; Staley, K.J.; White, J.A. Pyramidal cells accumulate chloride at seizure onset. Neurobiol. Dis. 2012, 47, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.S.; Artoni, P.; Landi, S.; Cozzolino, O.; Parra, R.; Pracucci, E.; Trovato, F.; Szczurkowska, J.; Luin, S.; Arosio, D.; et al. Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc. Natl. Acad. Sci. USA 2017, 114, E8770–E8779. [Google Scholar] [CrossRef] [Green Version]
- Raimondo, J.V.; Markram, H.; Akerman, C.J. Short-term ionic plasticity at GABAergic synapses. Front. Synaptic Neurosci. 2012, 4, 5. [Google Scholar] [CrossRef]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staley, K.J.; Proctor, W.R. Modulation of mammalian dendritic GABAA receptor function by the kinetics of Cl− and HCO3− transport. J. Physiol. 1999, 519, 693–712. [Google Scholar] [CrossRef] [PubMed]
- Jedlicka, P.; Backus, K.H. Inhibitory transmission, activity-dependent ionic changes and neuronal network oscillations. Physiol. Res. 2006, 55, 139–149. [Google Scholar] [PubMed]
- Wright, R.; Raimondo, J.V.; Akerman, C.J. Spatial and Temporal Dynamics in the Ionic Driving Force for GABA(A) Receptors. Neural Plast. 2011, 278395. [Google Scholar] [CrossRef]
- Doyon, N.; Prescott, S.A.; Castonguay, A.; Godin, A.G.; Kroger, H.; De Koninck, Y. Efficacy of Synaptic Inhibition Depends on Multiple, Dynamically Interacting Mechanisms Implicated in Chloride Homeostasis. PLoS Comput. Biol. 2011, 7, 9. [Google Scholar] [CrossRef]
- Mohapatra, N.; Tonnesen, J.; Vlachos, A.; Kuner, T.; Deller, T.; Nagerl, U.V.; Santamaria, F.; Jedlicka, P. Spines slow down dendritic chloride diffusion and affect short-term ionic plasticity of GABAergic inhibition. Sci. Rep. 2016, 6, 23196. [Google Scholar] [CrossRef] [PubMed]
- Buchin, A.; Chizhov, A.; Huberfeld, G.; Miles, R.; Gutkin, B.S. Reduced Efficacy of the KCC2 Cotransporter Promotes Epileptic Oscillations in a Subiculum Network Model. J. Neurosci. 2016, 36, 11619–11633. [Google Scholar] [CrossRef] [PubMed]
- Staley, K.J.; Soldo, B.L.; Proctor, W.R. Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 1995, 269, 977–981. [Google Scholar] [CrossRef]
- Sun, M.K.; Zhao, W.Q.; Nelson, T.J.; Alkon, D.L. Theta rhythm of hippocampal CA1 neuron activity: Gating by GABAergic synaptic depolarization. J. Neurophysiol. 2001, 85, 269–279. [Google Scholar] [CrossRef]
- Doyon, N.; Vinay, L.; Prescott, S.A.; De Koninck, Y. Chloride Regulation: A Dynamic Equilibrium Crucial for Synaptic Inhibition. Neuron 2016, 89, 1157–1172. [Google Scholar] [CrossRef]
- Thompson, S.M.; Gähwiler, B.H. Activity-dependent disinhibition. I. Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. J. Neurophysiol. 1989, 61, 501–511. [Google Scholar] [CrossRef]
- Doyon, N.; Prescott, S.A.; De Koninck, Y. Mild KCC2 Hypofunction Causes Inconspicuous Chloride Dysregulation that Degrades Neural Coding. Front. Cell. Neurosci. 2016, 9, 516. [Google Scholar] [CrossRef]
- Bernard, C.; Cossart, R.; Hirsch, J.C.; Esclapez, M.; Ben Ari, Y. What is GABAergic inhibition? How is it modified in epilepsy? Epilepsia 2000, 41, S90–S95. [Google Scholar] [CrossRef]
- Birke, G.; Draguhn, A. No Simple Brake—The Complex Functions of Inhibitory Synapses. Pharmacopsychiatry 2010, 43, S21–S31. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Cherubini, E.; Corradetti, R.; Gaiarsa, J.-L. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 1989, 416, 303–325. [Google Scholar] [CrossRef]
- Luhmann, H.J.; Prince, D.A. Postnatal maturation of the GABAergic system in rat neocortex. J. Neurophysiol. 1991, 65, 247–263. [Google Scholar] [CrossRef]
- Owens, D.F.; Boyce, L.H.; Davis, M.B.; Kriegstein, A.R. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. J. Neurosci. 1996, 16, 6414–6423. [Google Scholar] [CrossRef]
- Hanganu, I.L.; Kilb, W.; Luhmann, H.J. Functional Synaptic Projections onto Subplate Neurons in Neonatal Rat Somatosensory Cortex. J. Neurosci. 2002, 22, 7165–7176. [Google Scholar] [CrossRef]
- Achilles, K.; Okabe, A.; Ikeda, M.; Shimizu-Okabe, C.; Yamada, J.; Fukuda, A.; Luhmann, H.J.; Kilb, W. Kinetic properties of Cl uptake mediated by Na+-dependent K+-2Cl− cotransport in immature rat neocortical neurons. J. Neurosci. 2007, 27, 8616–8627. [Google Scholar] [CrossRef]
- Kirmse, K.; Kummer, M.; Kovalchuk, Y.; Witte, O.W.; Garaschuk, O.; Holthoff, K. GABA depolarizes immature neurons and inhibits network activity in the neonatal neocortex in vivo. Nat. Commun. 2015, 6, 7750. [Google Scholar] [CrossRef] [Green Version]
- Rohrbough, J.; Spitzer, N.C. Regulation of intracellular Cl− levels by Na(+)-dependent Cl− cotransport distinguishes depolarizing from hyperpolarizing GABAA receptor-mediated responses in spinal neurons. J. Neurosci. 1996, 16, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Yamada, J.; Okabe, A.; Toyoda, H.; Kilb, W.; Luhmann, H.J.; Fukuda, A. Cl− uptake promoting depolarizing GABA actions in immature rat neocortical neurones is mediated by NKCC1. J. Physiol. 2004, 557, 829–841. [Google Scholar] [CrossRef] [PubMed]
- Valeeva, G.; Tressard, T.; Mukhtarov, M.; Baude, A.; Khazipov, R. An Optogenetic Approach for Investigation of Excitatory and Inhibitory Network GABA Actions in Mice Expressing Channelrhodopsin-2 in GABAergic Neurons. J. Neurosci. 2016, 36, 5961–5973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolbaev, S.N.; Achilles, K.; Luhmann, H.J.; Kilb, W. Effect of depolarizing GABA(A)-mediated membrane responses on excitability of Cajal-Retzius cells in the immature rat neocortex. J. Neurophysiol. 2011, 106, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.F.; Kriegstein, A.R. Is there more to GABA than synaptic inhibition? Nat. Rev. Neurosci. 2002, 3, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Ben Ari, Y.; Khalilov, I.; Kahle, K.T.; Cherubini, E. The GABA Excitatory/Inhibitory Shift in Brain Maturation and Neurological Disorders. Neuroscientist 2012, 18, 467–486. [Google Scholar] [CrossRef] [PubMed]
- Luhmann, H.J.; Kirischuk, S.; Sinning, A.; Kilb, W. Early GABAergic circuitry in the cerebral cortex. Curr. Opin. Neurobiol. 2014, 26, 72–78. [Google Scholar] [CrossRef]
- Sipila, S.T.; Huttu, K.; Soltesz, I.; Voipio, J.; Kaila, K. Depolarizing GABA acts on intrinsically bursting pyramidal neurons to drive giant depolarizing potentials in the immature hippocampus. J. Neurosci. 2005, 25, 5280–5289. [Google Scholar] [CrossRef] [PubMed]
- Allene, C.; Cattani, A.; Ackman, J.B.; Bonifazi, P.; Aniksztejn, L.; Ben Ari, Y.; Cossart, R. Sequential Generation of Two Distinct Synapse-Driven Network Patterns in Developing Neocortex. J. Neurosci. 2008, 28, 12851–12863. [Google Scholar] [CrossRef] [Green Version]
- Chub, N.; O’Donovan, M.J. Post-episode depression of GABAergic transmission in spinal neurons of the chick embryo. J. Neurophysiol. 2001, 85, 2166–2176. [Google Scholar] [CrossRef]
- Lindsly, C.; Gonzalez-Islas, C.; Wenner, P. Activity Blockade and GABA(A) Receptor Blockade Produce Synaptic Scaling through Chloride Accumulation in Embryonic Spinal Motoneurons and Interneurons. PLoS ONE 2014, 9, e94559. [Google Scholar] [CrossRef] [PubMed]
- Kolbaev, S.N.; Luhmann, H.J.; Kilb, W. Activity-dependent scaling of GABAergic excitation by dynamic Cl− changes in Cajal-Retzius cells. Pflugers Arch. 2011, 461, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Islas, C.; Chub, N.; Garcia-Bereguiain, M.A.; Wenner, P. GABAergic synaptic scaling in embryonic motoneurons is mediated by a shift in the chloride reversal potential. J. Neurosci. 2010, 30, 13016–13020. [Google Scholar] [CrossRef] [Green Version]
- Khalilov, I.; Minlebaev, M.; Mukhtarov, M.; Khazipov, R. Dynamic Changes from Depolarizing to Hyperpolarizing GABAergic Actions during Giant Depolarizing Potentials in the Neonatal Rat Hippocampus. J. Neurosci. 2015, 35, 12635–12642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, A.; Jedlicka, P.; Luhmann, H.J.; Kilb, W. Giant Depolarizing Potentials Trigger Transient Changes in the Intracellular Cl− Concentration in CA3 Pyramidal Neurons of the Immature Mouse Hippocampus. Front. Cell. Neurosci. 2018, 12, 420. [Google Scholar] [CrossRef]
- Kowalski, J.; Gan, J.; Jonas, P.; Pernia-Andrade, A.J. Intrinsic membrane properties determine hippocampal differential firing pattern in vivo in anesthetized rats. Hippocampus 2016, 26, 668–682. [Google Scholar] [CrossRef]
- Behrens, C.J.; Ul Haq, R.; Liotta, A.; Anderson, M.L.; Heinemann, U. Nonspecific effects of the gap junction blocker mefloquine on fast hippocampal network oscillations in the adult rat in vitro. Neuroscience 2011, 192, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Bormann, J.; Hamill, O.P.; Sakmann, B. Mechanism of anion permeation through channels gated by glycine and gamma-aminobutyric acid in mouse cultured spinal neurones. J. Physiol. 1987, 385, 243–286. [Google Scholar] [CrossRef]
- Fatima-Shad, K.; Barry, P.H. Anion Permeation in GABA- and Glycine-Gated Channels of Mammalian Cultured Hippocampal Neurons. Proc. Biol. Sci. 1993, 253, 69–75. [Google Scholar]
- Ruusuvuori, E.; Li, H.; Huttu, K.; Palva, J.M.; Smirnov, S.; Rivera, C.; Kaila, K.; Voipio, J. Carbonic Anhydrase Isoform VII Acts as a Molecular Switch in the Development of Synchronous Gamma-Frequency Firing of Hippocampal CA1 Pyramidal Cells. J. Neurosci. 2004, 24, 2699–2707. [Google Scholar] [CrossRef] [Green Version]
- Kaila, K.; Voipio, J. Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature 1987, 330, 163–165. [Google Scholar] [CrossRef]
- Wang, G.J.; Randall, R.D.; Thayer, S.A. Glutamate-Induced Intracellular Acidification of Cultured Hippocampal-Neurons Demonstrates Altered Energy-Metabolism Resulting from Ca2+ Loads. J. Neurophysiol. 1994, 72, 2563–2569. [Google Scholar] [CrossRef]
- Kilb, W.; Schlue, W.R. Mechanism of the kainate-induced intracellular acidification in leech Retzius neurons. Brain Res. 1999, 824, 168–182. [Google Scholar] [CrossRef]
- Qian, N.; Sejnowski, T.J. When Is an Inhibitory Synapse Effective. Proc. Natl. Acad. Sci. USA 1990, 87, 8145–8149. [Google Scholar] [CrossRef]
- Kuner, T.; Augustine, G.J. A genetically encoded ratiometric indicator for chloride: Capturing chloride transients in cultured hippocampal neurons. Neuron 2000, 27, 447–459. [Google Scholar] [CrossRef]
- Kaila, K.; Ruusuvuori, E.; Seja, P.; Voipio, J.; Puskarjov, M. GABA actions and ionic plasticity in epilepsy. Curr. Opin. Neurobiol. 2014, 26, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, N.; Aksay, E.; Clancy, C.E. Computational modeling reveals dendritic origins of GABA(a)-mediated excitation in ca1 pyramidal neurons. PLoS ONE 2012, 7, e47250. [Google Scholar] [CrossRef]
- Dusterwald, K.M.; Currin, C.B.; Burman, R.J.; Akerman, C.J.; Kay, A.R.; Raimondo, J.V. Biophysical models reveal the relative importance of transporter proteins and impermeant anions in chloride homeostasis. Elife 2018, 7, e39575. [Google Scholar] [CrossRef]
- Luhmann, H.J.; Reiprich, R.A.; Hanganu, I.; Kilb, W. Cellular physiology of the neonatal rat cerebral cortex: Intrinsic membrane properties, sodium and calcium currents. J. Neurosci. Res. 2000, 62, 574–584. [Google Scholar] [CrossRef]
- Rheims, S.; Minlebaev, M.; Ivanov, A.; Represa, A.; Khazipov, R.; Holmes, G.L.; Ben-Ari, Y.; Zilberter, Y. Excitatory GABA in rodent developing neocortex in vitro. J. Neurophysiol. 2008, 100, 609–619. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Woodin, M.A.; Sernagor, E.; Cancedda, L.; Vinay, L.; Rivera, C.; Legendre, P.; Luhmann, H.J.; Bordey, A.; Wenner, P.; et al. Refuting the challenges of the developmental shift of polarity of GABA actions: GABA more exciting than ever! Front. Cell. Neurosci. 2012, 6, 35. [Google Scholar] [CrossRef]
- Kaila, K.; Lamsa, K.; Smirnov, S.; Taira, T.; Voipio, J. Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J. Neurosci. 1997, 17, 7662–7672. [Google Scholar] [CrossRef] [PubMed]
- Dallwig, R.; Deitmer, J.W.; Backus, K.H. On the mechanism of GABA-induced currents in cultured rat cortical neurons. Pflugers Arch. 1999, 437, 289–297. [Google Scholar] [CrossRef]
- Sun, M.; Dahl, D.; Alkon, D.L. Heterosynaptic transformation of GABAergic gating in the hippocampus and effects of carbonic anhydrase inhibition. J. Pharmacol. Exp. Ther. 2001, 296, 811–817. [Google Scholar] [PubMed]
- Rivera, C.; Voipio, J.; Kaila, K. Two developmental switches in GABAergic signalling: The K+- Cl− cotransporter KCC2 and carbonic anhydrase CAVII. J. Physiol. 2005, 562, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Earnhardt, J.N.; Qian, M.Z.; Tu, C.K.; Lakkis, M.M.; Bergenhem, N.C.H.; Laipis, P.J.; Tashian, R.E.; Silverman, D.N. The catalytic properties of murine carbonic anhydrase VII. Biochemistry 1998, 37, 10837–10845. [Google Scholar] [CrossRef] [PubMed]
- Kaila, K. Ionic basis of GABAA receptor channel function in the nervous system. Prog. Neurobiol. 1994, 42, 489–537. [Google Scholar] [CrossRef]
- Doischer, D.; Hosp, J.A.; Yanagawa, Y.; Obata, K.; Jonas, P.; Vida, I.; Bartos, M. Postnatal Differentiation of Basket Cells from Slow to Fast Signaling Devices. J. Neurosci. 2008, 28, 12956–12968. [Google Scholar] [CrossRef] [Green Version]
- Kilb, W.; Luhmann, H.J. Spontaneous GABAergic postsynaptic currents in Cajal-Retzius cells in neonatal rat cerebral cortex. Eur. J. Neurosci. 2001, 13, 1387–1390. [Google Scholar] [CrossRef]
- Kubota, Y. Untangling GABAergic wiring in the cortical microcircuit. Curr. Opin. Neurobiol. 2014, 26, 7–14. [Google Scholar] [CrossRef]
- Misgeld, U.; Deisz, R.A.; Dodt, H.U.; Lux, H.D. The role of chloride transport in postsynaptic inhibition of hippocampal neurons. Science 1986, 232, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Islas, C.; Chub, N.; Wenner, P. NKCC1 and AE3 appear to accumulate chloride in embryonic motoneurons. J. Neurophysiol. 2009, 101, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Kakazu, Y.; Uchida, S.; Nakagawa, T.; Akaike, N.; Nabekura, J. Reversibility and cation selectivity of the K+-Cl− cotransport in rat central neurons. J. Neurophysiol. 2000, 84, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Kahle, K.T.; Staley, K.J.; Nahed, B.V.; Gamba, G.; Hebert, S.C.; Lifton, R.P.; Mount, D.B. Roles of the cation-chloride cotransporters in neurological disease. Nat. Clin. Pract. Neurol. 2008, 4, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Khirug, S.; Ahmad, F.; Puskarjov, M.; Afzalov, R.; Kaila, K.; Blaesse, P. A single seizure episode leads to rapid functional activation of KCC2 in the neonatal rat hippocampus. J. Neurosci. 2010, 30, 12028–12035. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Furukawa, T.; Kumada, T.; Yamada, J.; Wang, T.Y.; Inoue, R.; Fukuda, A. Taurine Inhibits K+-Cl− Cotransporter KCC2 to Regulate Embryonic Cl− Homeostasis via With-no-lysine (WNK) Protein Kinase Signaling Pathway. J. Biol. Chem. 2012, 287, 20839–20850. [Google Scholar] [CrossRef]
- Russell, J.M. Sodium-potassium-chloride cotransport. Physiol. Rev. 2000, 80, 211–276. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E.; Austin, T.M. Kinase regulation of Na+-K+-2Cl− cotransport in primary afferent neurons. J. Physiol. 2010, 588, 3365–3373. [Google Scholar] [CrossRef]
- Bar-Yehuda, D.; Korngreen, A. Space-clamp problems when voltage clamping neurons expressing voltage-gated conductances. J. Neurophysiol. 2008, 99, 1127–1136. [Google Scholar] [CrossRef]
- Backus, K.H.; Deitmer, J.W.; Friauf, E. Glycine-activated currents are changed by coincident membrane depolarization in developing rat auditory brainstem neurones. J. Physiol. 1998, 507, 783–794. [Google Scholar] [CrossRef] [Green Version]
- Ellis, R.J.; Minton, A.P. Cell biology—Join the crowd. Nature 2003, 425, 27–28. [Google Scholar] [CrossRef] [PubMed]
- McDougal, R.A.; Morse, T.M.; Carnevale, T.; Marenco, L.; Wang, R.; Migliore, M.; Miller, P.L.; Shepherd, G.M.; Hines, M.L. Twenty years of ModelDB and beyond: Building essential modeling tools for the future of neuroscience. J. Comput. Neurosci. 2017, 42, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, K.; Armstrong, W.E. A versatile means of intracellular labeling: Injection of biocytin and its detection with avidin conjugates. J. Neurosci. Meth. 1988, 25, 1–11. [Google Scholar] [CrossRef]
- Schröder, R.; Luhmann, H.J. Morphology, electrophysiology and pathophysiology of supragranular neurons in rat primary somatosensory cortex. Eur. J. Neurosci. 1997, 9, 163–176. [Google Scholar] [CrossRef] [PubMed]
- De Schutter, E.; Smolen, P. Calcium Dynamics in Large Neuronal Models. In Methods in Neuronal Modeling; Koch, C., Segev, I., Eds.; MIT Press: Cambridge, MA, USA, 1998; pp. 211–250. [Google Scholar]
- De Schutter, E. Modeling intracellular calcium dynamics. In Computational Modeling Methods for Neuroscientists; MIT Press: Cambridge, MA, USA, 2010; pp. 93–105. [Google Scholar]
- Mohapatra, N.; Deans, H.; Santamaria, F.; Jedlicka, P. Modeling Ion Concentrations. In Encyclopedia of Computational Neuroscience; Jaeger, D., Jung, R., Eds.; Springer: New York, NY, USA, 2015. [Google Scholar] [CrossRef]
- Hines, M.L.; Carnevale, N.T. Expanding NEURON’s repertoire of mechanisms with NMODL. Neural Comput. 2000, 12, 995–1007. [Google Scholar] [CrossRef]
- Santhakumar, V.; Aradi, I.; Soltesz, I. Role of mossy fiber sprouting and mossy cell loss in hyperexcitability: A network model of the dentate gyrus incorporating cell types and axonal topography. J. Neurophysiol. 2005, 93, 437–453. [Google Scholar] [CrossRef] [PubMed]
- Ruusuvuori, E.; Kirilkin, I.; Pandya, N.; Kaila, K. Spontaneous network events driven by depolarizing GABA action in neonatal hippocampal slices are not attributable to deficient mitochondrial energy metabolism. J. Neurosci. 2010, 30, 15638–15642. [Google Scholar] [CrossRef]
- Mitchell, R.A.; Herbert, D.A.; Carman, C.T. Acid-Base Constants and Temperature Coefficients for Cerebrospinal Fluid. J. Appl. Physiol. 1965, 20, 27–30. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombardi, A.; Jedlicka, P.; Luhmann, H.J.; Kilb, W. Interactions between Membrane Resistance, GABA-A Receptor Properties, Bicarbonate Dynamics and Cl−-Transport Shape Activity-Dependent Changes of Intracellular Cl− Concentration. Int. J. Mol. Sci. 2019, 20, 1416. https://doi.org/10.3390/ijms20061416
Lombardi A, Jedlicka P, Luhmann HJ, Kilb W. Interactions between Membrane Resistance, GABA-A Receptor Properties, Bicarbonate Dynamics and Cl−-Transport Shape Activity-Dependent Changes of Intracellular Cl− Concentration. International Journal of Molecular Sciences. 2019; 20(6):1416. https://doi.org/10.3390/ijms20061416
Chicago/Turabian StyleLombardi, Aniello, Peter Jedlicka, Heiko J. Luhmann, and Werner Kilb. 2019. "Interactions between Membrane Resistance, GABA-A Receptor Properties, Bicarbonate Dynamics and Cl−-Transport Shape Activity-Dependent Changes of Intracellular Cl− Concentration" International Journal of Molecular Sciences 20, no. 6: 1416. https://doi.org/10.3390/ijms20061416