Energy Landscapes of Ligand Motion Inside the Tunnel-Like Cavity of Lipid Transfer Proteins: The Case of the Pru p 3 Allergen

 , ,
, ,
Abstract
:1. Introduction
2. Results
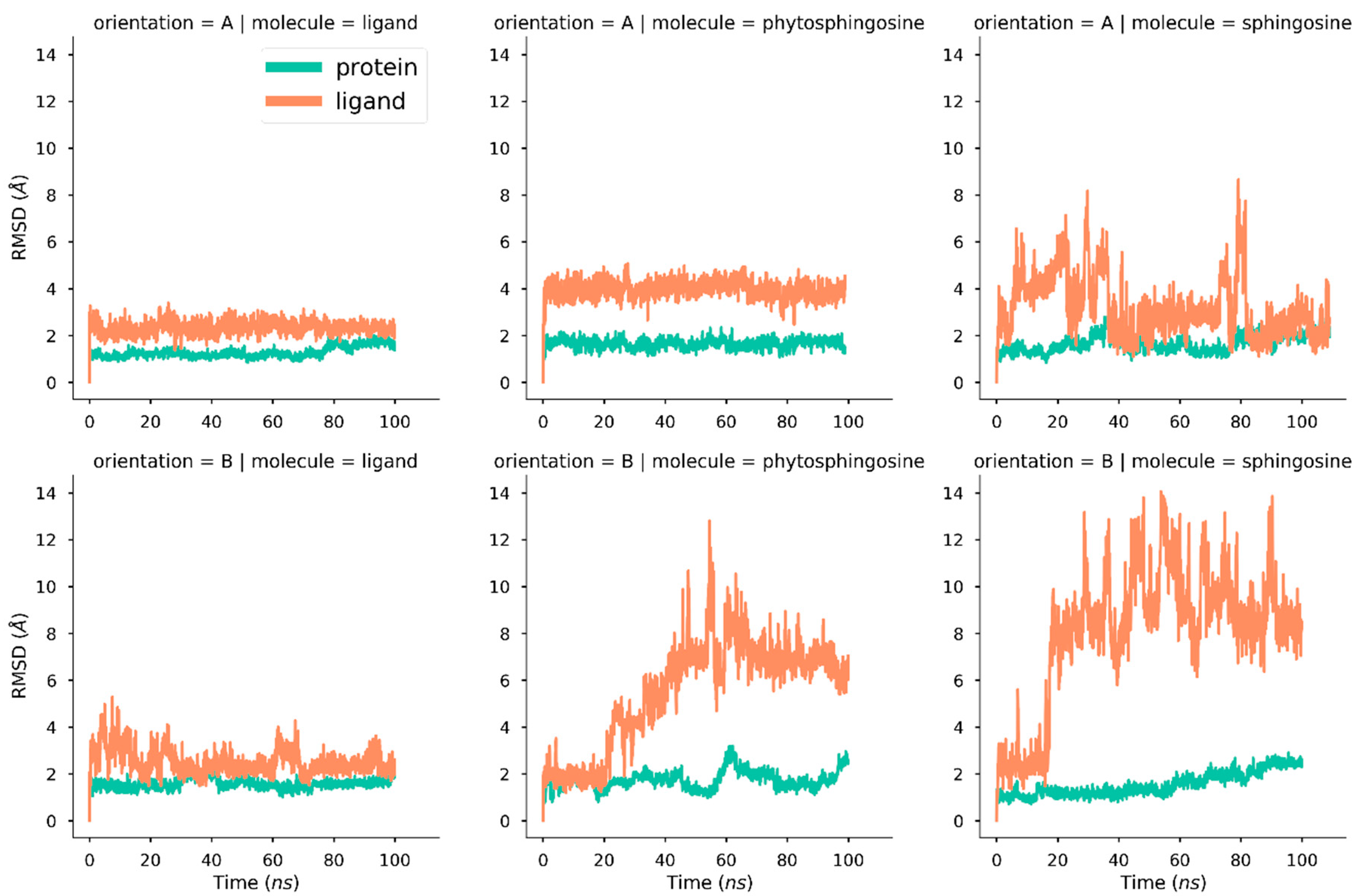
2.1. Molecular Dynamics
2.2. Protein–Ligand Binding Free Energy
2.3. Collective Variables of Ligand–Protein Motion
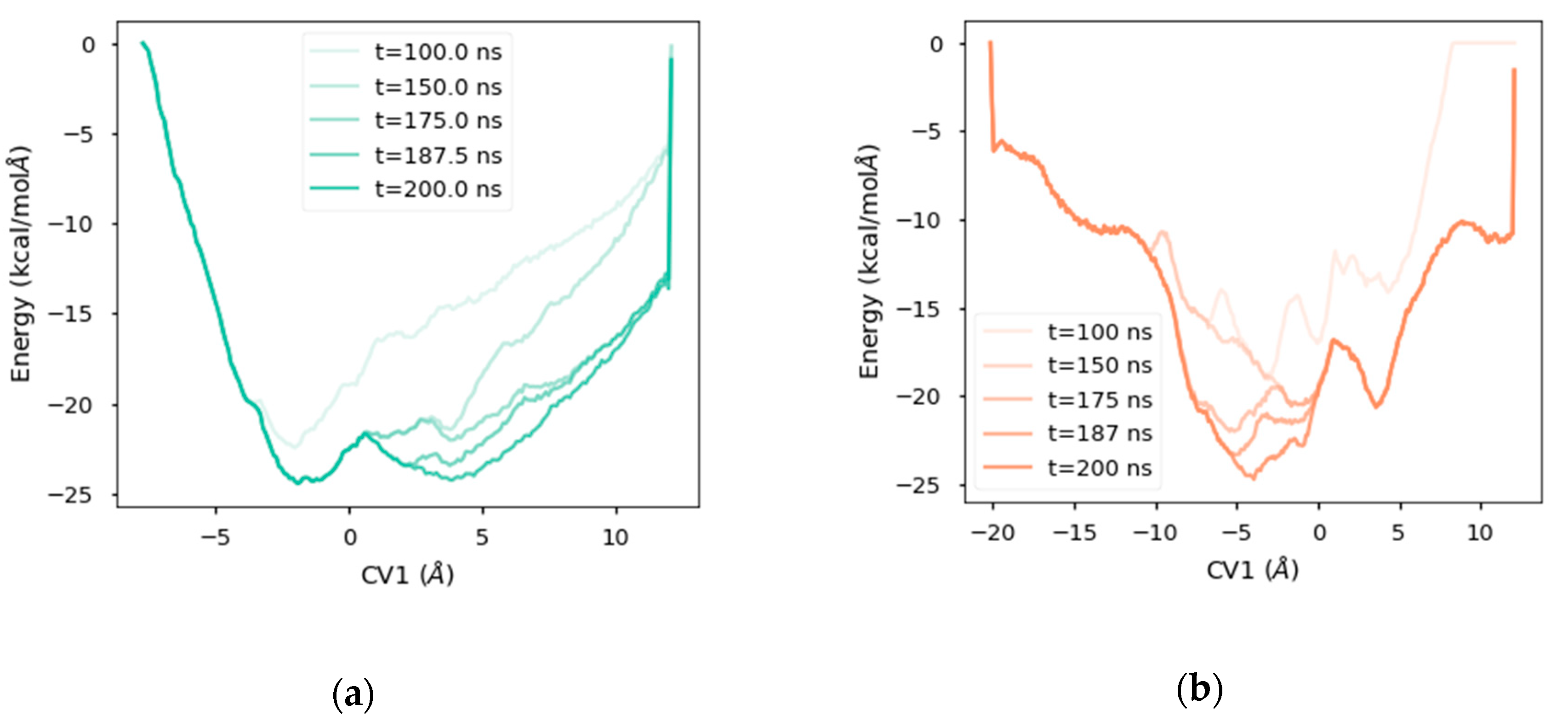
2.4. Free Energy Landscape
2.5. Analysis of the Diffusion Process
3. Discussion
4. Materials and Methods
4.1. Protein Structure
4.2. Binding Poses of Ligands
4.3. Force Field Parametrization
4.4. Molecular Dynamics
4.5. Map of Interacting Residues
4.6. Free Energy Estimation
4.7. Collective Variables Setting
4.8. Metadynamics
4.9. Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CV | Collective variable |
| LTP | Lipid transfer protein |
| MD | Molecular dynamics |
| PCA | Principal components analysis |
| RMSD | Root mean squares deviation |
| RMSF | Root mean squares fluctuation |
Appendix A
References
- Pawankar, R. White Book on Allergy; World Allergy Organization: Milwaukee, WI, USA, 2013; ISBN 0002-9378. [Google Scholar]
- Rudders, S.A.; Arias, S.A.; Camargo, C.A. Trends in hospitalizations for food-induced anaphylaxis in US children, 2000–2009. J. Allergy Clin. Immunol. 2014, 134, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.; Mugford, M.; Voordouw, J.; Cornelisse-Vermaat, J.; Antonides, G.; de la Hoz Caballer, B.; Cerecedo, I.; Zamora, J.; Rokicka, E.; Jewczak, M.; et al. Health sector costs of self-reported food allergy in Europe: A patient-based cost of illness study. Eur. J. Public Health 2013, 23, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Sampson, H.A.; O’Mahony, L.; Burks, A.W.; Plaut, M.; Lack, G.; Akdis, C.A. Mechanisms of food allergy. J. Allergy Clin. Immunol. 2018, 141, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-Sommergruber, K. Plant allergens and pathogenesis-related proteins. Int. Arch. Allergy Immunol. 2000, 122, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Bublin, M.; Eiwegger, T.; Breiteneder, H. Do lipids influence the allergic sensitization process? J. Allergy Clin. Immunol. 2014, 134, 521–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, W.R. Allergen ligands in the initiation of allergic sensitization. Curr. Allergy Asthma Rep. 2014, 14, 432. [Google Scholar] [CrossRef] [PubMed]
- Lerche, M.H.; Poulsen, F.M. Solution structure of barley lipid transfer protein complexed with palmitate. Two different binding modes of palmitate in the homologous maize and barley nonspecific lipid transfer proteins. Protein Sci. 1998, 7, 2490–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquato, N.; Berni, R.; Folli, C.; Folloni, S.; Cianci, M.; Pantano, S.; Helliwell, J.R.; Zanotti, G. Crystal structure of peach Pru p 3, the prototypic member of the family of plant non-specific lipid transfer protein pan-allergens. J. Mol. Biol. 2006, 356, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Shenkarev, Z.O.; Melnikova, D.N.; Finkina, E.I.; Sukhanov, S.V.; Boldyrev, I.A.; Gizatullina, A.K.; Mineev, K.S.; Arseniev, A.S.; Ovchinnikova, T.V. Ligand binding properties of the lentil lipid transfer protein: Molecular insight into the possible mechanism of lipid uptake. Biochemistry 2017, 56, 1785–1796. [Google Scholar] [CrossRef] [PubMed]
- Charvolin, D.; Douliez, J.P.; Marion, D.; Cohen-Addad, C.; Pebay-Peyroula, E. The crystal structure of a wheat nonspecific lipid transfer protein (ns-LTP1) complexed with two molecules of phospholipid at 2.1 A resolution. Eur. J. Biochem. 1999, 264, 562–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, T.A.; Blomqvist, K.; Edqvist, J. Lipid transfer proteins: Classification, nomenclature, structure, and function. Planta 2016, 244, 971–997. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, S.U.; Alexeev, Y.; Johnson, P.E.; Rigby, N.M.; Mackie, A.R.; Dhaliwal, B.; Mills, E.N.C. Ligand binding to an allergenic lipid transfer protein enhances conformational flexibility resulting in an increase in susceptibility to gastroduodenal proteolysis. Sci. Rep. 2016, 6, 30279. [Google Scholar] [CrossRef] [PubMed]
- Pacios, L.F.; Gómez-Casado, C.; Tordesillas, L.; Palacín, A.; Sánchez-Monge, R.; Díaz-Perales, A. Computational study of ligand binding in lipid transfer proteins: Structures, interfaces, and free energies of protein-lipid complexes. J. Comput. Chem. 2012, 33, 1831–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina, A.; García-Olmedo, F. Developmental and pathogen-induced expression of three barley genes encoding lipid transfer proteins. Plant J. 1993, 4, 983–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pato, C.; Le Borgne, M.; Le Baut, G.; Le Pape, P.; Marion, D.; Douliez, J.-P. Potential application of plant lipid transfer proteins for drug delivery. Biochem. Pharmacol. 2001, 62, 555–560. [Google Scholar] [CrossRef]
- Choi, E.J.; Mao, J.; Mayo, S.L. Computational design and biochemical characterization of maize nonspecific lipid transfer protein variants for biosensor applications. Protein Sci. 2007, 16, 582–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubells-Baeza, N.; Gómez-Casado, C.; Tordesillas, L.; Ramírez-Castillejo, C.; Garrido-Arandia, M.; González-Melendi, P.; Herrero, M.; Pacios, L.F.; Díaz-Perales, A. Identification of the ligand of Pru p 3, a peach LTP. Plant Mol. Biol. 2017, 94, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Markham, J.E.; Li, J.; Cahoon, E.B.; Jaworski, J.G. Separation and identification of major plant sphingolipid classes from leaves. J. Biol. Chem. 2006, 281, 22684–22694. [Google Scholar] [CrossRef]
- Tordesillas, L.; Cubells-Baeza, N.; Gómez-Casado, C.; Berin, C.; Esteban, V.; Barcik, W.; O’Mahony, L.; Ramirez, C.; Pacios, L.F.; Garrido-Arandia, M.; et al. Mechanisms underlying induction of allergic sensitization by Pru p 3. Clin. Exp. Allergy 2017, 47, 1398–1408. [Google Scholar] [CrossRef]
- Lai, Y.T.; Cheng, C.S.; Liu, Y.N.; Liu, Y.J.; Lyu, P.C. Effects of ligand binding on the dynamics of rice nonspecific lipid transfer protein 1: A model from molecular simulations. Proteins Struct. Funct. Genet. 2008, 72, 1189–1198. [Google Scholar] [CrossRef]
- Smith, L.J.; van Gunsteren, W.F.; Allison, J.R. Multiple binding modes for palmitate to barley lipid transfer protein facilitated by the presence of proline 12. Protein Sci. 2013, 22, 56–64. [Google Scholar] [CrossRef]
- Shi, Z.; Wang, Z.; Xu, H.; Tian, Y.; Li, X.; Bao, J.; Sun, S.; Yue, B. Modeling, docking and dynamics simulations of a non-specific lipid transfer protein from Peganum harmala L. Comput. Biol. Chem. 2013, 47, 56–65. [Google Scholar] [CrossRef]
- Tousheh, M.; Miroliaei, M.; Asghar Rastegari, A.; Ghaedi, K.; Esmaeili, A.; Matkowski, A. Computational evaluation on the binding affinity of non-specific lipid-transfer protein-2 with fatty acids. Comput. Biol. Med. 2013, 43, 1732–1738. [Google Scholar] [CrossRef]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Reports Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Gervasio, F.L.; Laio, A.; Parrinello, M. Flexible docking in solution using supporting information. Support. Inf. 2004, 127, 1–5. [Google Scholar]
- Tiwary, P.; Limongelli, V.; Salvalaglio, M.; Parrinello, M. Kinetics of protein–ligand unbinding: Predicting pathways, rates, and rate-limiting steps. Proc. Natl. Acad. Sci. USA 2015, 112, E386–E391. [Google Scholar] [CrossRef]
- Clark, A.J.; Tiwary, P.; Borrelli, K.; Feng, S.; Miller, E.B.; Abel, R.; Friesner, R.A.; Berne, B.J. Prediction of protein-ligand binding poses via a combination of induced fit docking and metadynamics simulations. J. Chem. Theory Comput. 2016, 12, 2990–2998. [Google Scholar] [CrossRef]
- Casasnovas, R.; Limongelli, V.; Tiwary, P.; Carloni, P.; Parrinello, M. Unbinding kinetics of a p38 MAP kinase type II inhibitor from metadynamics simulations. J. Am. Chem. Soc. 2017, 139, 4780–4788. [Google Scholar] [CrossRef]
- Provasi, D.; Bortolato, A.; Filizola, M. Exploring molecular mechanisms of ligand recognition by opioid receptors with metadynamics. Biochemistry 2009, 48, 10020–10029. [Google Scholar] [CrossRef]
- Cao, Z.; Bian, Y.; Hu, G.; Zhao, L.; Kong, Z.; Yang, Y.; Wang, J.; Zhou, Y. Bias-exchange metadynamics simulation of membrane permeation of 20 amino acids. Int. J. Mol. Sci. 2018, 19, 885. [Google Scholar] [CrossRef]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008, 100, 1–4. [Google Scholar] [CrossRef]
- Rydzewski, J.; Nowak, W. Ligand diffusion in proteins via enhanced sampling in molecular dynamics. Phys. Life Rev. 2017, 22–23, 58–74. [Google Scholar] [CrossRef]
- Michaelson, L.V.; Napier, J.A.; Molino, D.; Faure, J.-D. Plant sphingolipids: Their importance in cellular organization and adaption. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Bidon-Chanal, A.; Luque, F.J.; Barril, X. MDpocket: Open-source cavity detection and characterization on molecular dynamics trajectories. Bioinformatics 2011, 27, 3276–3285. [Google Scholar] [CrossRef] [PubMed]
- Craig, I.R.; Pfleger, C.; Gohlke, H.; Essex, J.W.; Spiegel, K. Pocket-space maps to identify novel binding-site conformations in proteins. J. Chem. Inf. Model. 2011, 51, 2666–2679. [Google Scholar] [CrossRef] [PubMed]
- Lerche, M.H.; Kragelund, B.B.; Bech, L.M.; Poulsen, F.M. Barley lipid-transfer protein complexed with palmitoyl CoA: The structure reveals a hydrophobic binding site that can expand to fit both large and small lipid-like ligands. Structure 1997, 5, 291–306. [Google Scholar] [CrossRef]
- Edstam, M.M.; Viitanen, L.; Salminen, T.A.; Edqvist, J. Evolutionary history of the non-specific lipid transfer proteins. Mol. Plant 2011, 4, 947–964. [Google Scholar] [CrossRef]
- García-Olmedo, F.; Molina, A.; Alamillo, J.M.; Rodríguez-Palenzuéla, P. Plant defense peptides. Biopolymers 1998, 47, 479–491. [Google Scholar] [CrossRef]
- Cammue, B.P.; Thevissen, K.; Hendriks, M.; Eggermont, K.; Goderis, I.J.; Proost, P.; van Damme, J.; Osborn, R.W.; Guerbette, F.; Kader, J.C. A potent antimicrobial protein from onion seeds showing sequence homology to plant lipid transfer proteins. Plant Physiol. 1995, 109, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Ooi, L.S.M.; Tian, L.; Su, M.; Ho, W.-S.; Sun, S.S.M.; Chung, H.-Y.; Wong, H.N.C.; Ooi, V.E.C. Isolation, characterization, molecular cloning and modeling of a new lipid transfer protein with antiviral and antiproliferative activities from Narcissus tazetta. Peptides 2008, 29, 2101–2109. [Google Scholar] [CrossRef]
- Cameron, K.D.; Teece, M.A.; Smart, L.B. Increased accumulation of cuticular wax and expression of lipid transfer protein in response to periodic drying events in leaves of tree tobacco. Plant Physiol. 2006, 140, 176–183. [Google Scholar] [CrossRef]
- Park, S.Y.; Jauh, G.Y.; Mollet, J.C.; Eckard, K.J.; Nothnagel, E.A.; Walling, L.L.; Lord, E.M. A lipid transfer-like protein is necessary for lily pollen tube adhesion to an in vitro stylar matrix. Plant Cell 2000, 12, 151–164. [Google Scholar] [CrossRef]
- Crimi, M.; Astegno, A.; Zoccatelli, G.; Esposti, M.D. Pro-apoptotic effect of maize lipid transfer protein on mammalian mitochondria. Arch. Biochem. Biophys. 2006, 445, 65–71. [Google Scholar] [CrossRef]
- Rudrappa, T.; Biedrzycki, M.L.; Kunjeti, S.G.; Donofrio, N.M.; Czymmek, K.J.; Paré, P.W.; Bais, H.P. The rhizobacterial elicitor acetoin induces systemic resistance in Arabidopsis thaliana. Commun. Integr. Biol. 2010, 3, 130–138. [Google Scholar] [CrossRef]
- Harrison, P.J.; Dunn, T.M.; Campopiano, D.J. Sphingolipid biosynthesis in man and microbes. Nat. Prod. Rep. 2018, 35, 921–954. [Google Scholar] [CrossRef]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: From synthesis to breakdown. Adv. Exp. Med. Biol. 2010, 688, 1–23. [Google Scholar]
- Höglinger, D.; Haberkant, P.; Aguilera-Romero, A.; Riezman, H.; Porter, F.D.; Platt, F.M.; Galione, A.; Schultz, C. Intracellular sphingosine releases calcium from lysosomes. Elife 2015, 4, e10616. [Google Scholar] [CrossRef]
- Branduardi, D.; Gervasio, F.L.; Parrinello, M. From A to B in free energy space. J. Chem. Phys. 2007, 126, 054103. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.-J.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem. 2017, 38, 1114–1124. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins †. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Liu, H.; Hou, T.; Ca, F.E. A tool for binding affinity prediction using end-point free energy methods. Bioinformatics 2016, 32, 2216–2218. [Google Scholar] [CrossRef]
- Glykos, N.M. Software news and updates carma: A molecular dynamics analysis program. J. Comput. Chem. 2006, 27, 1765–1768. [Google Scholar] [CrossRef]
- Fiorin, G.; Klein, M.L.; Hénin, J. Using collective variables to drive molecular dynamics simulations. Mol. Phys. 2013, 111, 3345–3362. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33, 27–28. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Orientation | ΔGbind (kcal/mol) | ΔH (kcal/mol) | TΔS (kcal/mol) |
|---|---|---|---|---|
| Natural Ligand | A | −7.27 ± 0.59 | −18.63 ± 0.68 | −11.36 ± 0.23 |
| Natural Ligand | B | −11.15 ± 0.58 | −23.03 ± 0.49 | −11.88 ± 0.26 |
| Phytosphingosine | A | −10.20 ± 0.41 | −21.73 ± 0.38 | 11.53 ± 0.19 |
| Phytosphingosine | B | −3.33 ± 0.49 | −16.07 ± 0.47 | −12.75 ± 0.18 |
| Sphingosine | A | −6.09 ± 0.59 | −19.02 ± 0.59 | −12.93 ± 0.20 |
| Sphingosine | B | −9.36 ± 0.33 | −22.48 ± 0.27 | −13.12 ± 0.23 |
| Type of Bias | Variable | Parameters |
|---|---|---|
| Harmonic wall | CV2 | K = 40.0 kcal/Å·mol, s0− = −20 Å, s0+ = 12.5 Å |
| Harmonic wall | CV1 | K = 40.0 kcal/Å·mol, s0− = 0 Å, s0+ = 9.0 Å |
| WT-Metadynamics | CV1 | H = 0.1 kcal/Å·mol, δ = 0.05 Å, Δ T = 4500 K, τG = 1000 fs |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuevas-Zuviría, B.; Garrido-Arandia, M.; Díaz-Perales, A.; Pacios, L.F. Energy Landscapes of Ligand Motion Inside the Tunnel-Like Cavity of Lipid Transfer Proteins: The Case of the Pru p 3 Allergen. Int. J. Mol. Sci. 2019, 20, 1432. https://doi.org/10.3390/ijms20061432
Cuevas-Zuviría B, Garrido-Arandia M, Díaz-Perales A, Pacios LF. Energy Landscapes of Ligand Motion Inside the Tunnel-Like Cavity of Lipid Transfer Proteins: The Case of the Pru p 3 Allergen. International Journal of Molecular Sciences. 2019; 20(6):1432. https://doi.org/10.3390/ijms20061432
Chicago/Turabian StyleCuevas-Zuviría, Bruno, María Garrido-Arandia, Araceli Díaz-Perales, and Luis F. Pacios. 2019. "Energy Landscapes of Ligand Motion Inside the Tunnel-Like Cavity of Lipid Transfer Proteins: The Case of the Pru p 3 Allergen" International Journal of Molecular Sciences 20, no. 6: 1432. https://doi.org/10.3390/ijms20061432