Analysis of 50 Neurodegenerative Genes in Clinically Diagnosed Early-Onset Alzheimer’s Disease

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Novel and Known AD Pathogenic Variants
2.2. Known FTD Pathogenic Variant
2.3. Known PD Pathogenic Variants
2.4. Other Novel and Known Dementia Variants
2.5. In Silico Gene/Protein Functional Interaction Network
3. Discussion
4. Materials and Methods
4.1. Study Samples
4.2. Selection of Candidate Genes, Variants, and Analysis
4.3. Ion Torrent PGM Sequencing and NGS Processing
4.4. In Silico Gene/Protein Functional Interaction Network
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S. Clinical genetic strategies for early onset neurodegenerative diseases. Mol. Cell. Toxicol. 2018, 14, 123–142. [Google Scholar] [CrossRef]
- Arneson, D.; Zhang, Y.; Yang, X.; Narayanan, M. Shared mechanisms among neurodegenerative diseases: From genetic factors to gene networks. J. Genet. 2018, 97, 795–806. [Google Scholar] [CrossRef]
- Hagenaars, S.P.; Radaković, R.; Crockford, C.; Fawns-Ritchie, C.; International FTD-Genomics Consortium; Harris, S.E.; Gale, C.R.; Deary, I.J. Genetic risk for neurodegenerative disorders, and its overlap with cognitive ability and physical function. PLoS ONE 2018, 13, e0198187. [Google Scholar] [CrossRef]
- Van Giau, V.; An, S.S.A.; Bagyinszky, E.; Kim, S. Gene panels and primers for next generation sequencing studies on neurodegenerative disorders. Mol. Cell. Toxicol. 2015, 11, 89–143. [Google Scholar] [CrossRef]
- Santiago, J.A.; Bottero, V.; Potashkin, J.A. Dissecting the Molecular Mechanisms of Neurodegenerative Diseases through Network Biology. Front. Aging Neurosci. 2017, 9, 166. [Google Scholar] [CrossRef]
- Lill, C.M.; Bertram, L. Towards unveiling the genetics of neurodegenerative diseases. Semin. Neurol. 2011, 31, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2013, 9, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Giau, V.V.; Vo, T.K. Current advances in transdermal delivery of drugs for Alzheimer’s disease. Indian J. Pharmacol. 2017, 49, 145–154. [Google Scholar]
- Bagyinszky, E.; Giau, V.V.; Shim, K.; Suk, K.; An, S.S.A.; Kim, S. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J. Neurol. Sci. 2017, 376, 242–254. [Google Scholar] [CrossRef]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Van Giau, V.; Senanarong, V.; Bagyinszky, E.; Limwongse, C.; An, S.S.A.; Kim, S. Identification of a novel mutation in APP gene in a Thai subject with early-onset Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2018, 14, 3015–3023. [Google Scholar] [CrossRef]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Wang, M.J.; Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. Novel PSEN1 p.Gly417Ala mutation in a Korean patient with early-onset Alzheimer’s disease with parkinsonism. Neurobiol. Aging 2018, 72, 188.e13–188.e17. [Google Scholar] [CrossRef]
- Park, J.; An, S.S.A.; Giau, V.V.; Shim, K.; Youn, Y.C.; Bagyinszky, E.; Kim, S. Identification of a novel PSEN1 mutation (Leu232Pro) in a Korean patient with early-onset Alzheimer’s disease and a family history of dementia. Neurobiol. Aging 2017, 56, 212.e11–212.e17. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Pyun, J.-M.; Bagyinszky, E.; An, S.S.A.; Kim, S. A pathogenic PSEN2 p.His169Asn mutation associated with early-onset Alzheimer’s disease. Clin. Interv. Aging 2018, 13, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Monsell, S.E.; Phillips, L.E.; Kukull, W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J. Neuropathol. Exp. Neurol. 2012, 71, 266–273. [Google Scholar] [CrossRef]
- Van Giau, V.; An, S.S.A. Optimization of specific multiplex DNA primers to detect variable CLU genomic lesions in patients with Alzheimer’s disease. BioChip J. 2015, 9, 278–284. [Google Scholar] [CrossRef]
- Lord, J.; Lu, A.J.; Cruchaga, C. Identification of rare variants in Alzheimer’s disease. Front. Genet. 2014, 5, 369. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, J.M.; Hinz, W.; Rearick, T.M.; Schultz, J.; Mileski, W.; Davey, M.; Leamon, J.H.; Johnson, K.; Milgrew, M.J.; Edwards, M.; et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature 2011, 475, 348–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallon, D.; Rousseau, S.; Rovelet-Lecrux, A.; Quillard-Muraine, M.; Guyant-Marechal, L.; Martinaud, O.; Pariente, J.; Puel, M.; Rollin-Sillaire, A.; Pasquier, F.; et al. The French series of autosomal dominant early onset Alzheimer’s disease cases: Mutation spectrum and cerebrospinal fluid biomarkers. J. Alzheimer’s Dis. 2012, 30, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Gu, X.; Wei, J.; Jiao, B.; Zhou, L.; Zhou, Y.; Weng, L.; Yan, X.; Tang, B.; Xu, J.; et al. Analyses MAPT, GRN, and C9orf72 mutations in Chinese patients with frontotemporal dementia. Neurobiol. Aging 2016, 46, 235.e11–235.e15. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Theuns, J.; Van Broeckhoven, C. Locus-specific mutation databases for neurodegenerative brain diseases. Hum. Mutat. 2012, 33, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Nuytemans, K.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Zabetian, C.P.; Lauricella, C.J.; Tsuang, D.W.; Leverenz, J.B.; Schellenberg, G.D.; Payami, H. Analysis of the LRRK2 G2019S mutation in Alzheimer Disease. Arch. Neurol. 2006, 63, 156–157. [Google Scholar] [CrossRef]
- Zhao, Y.; Ho, P.; Yih, Y.; Chen, C.; Lee, W.L.; Tan, E.K. LRRK2 variant associated with Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1990–1993. [Google Scholar] [CrossRef]
- Tan, E.K.; Peng, R.; Teo, Y.Y.; Tan, L.C.; Angeles, D.; Ho, P.; Chen, M.L.; Lin, C.H.; Mao, X.Y.; Chang, X.L.; et al. Multiple LRRK2 variants modulate risk of Parkinson disease: A Chinese multicenter study. Hum. Mutat. 2010, 31, 561–568. [Google Scholar]
- Ng, A.S.L.; Ng, E.Y.L.; Tan, Y.J.; Kandiah, N.; Zhou, J.; Hameed, S.; Ting, S.K.S.; Tan, E.K. Case-control analysis of leucine-rich repeat kinase 2 protective variants in Alzheimer’s disease. Neurobiol. Aging 2018, 64, 157.e7–157.e9. [Google Scholar] [CrossRef]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Kober, D.L.; Alexander-Brett, J.M.; Karch, C.M.; Cruchaga, C.; Colonna, M.; Holtzman, M.J.; Brett, T.J. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. eLife 2016, 5, e20391. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.C.; Carrasquillo, M.M.; Benitez, B.A.; Skorupa, T.; Carrell, D.; Patel, D.; Lincoln, S.; Krishnan, S.; Kachadoorian, M.; Reitz, C.; et al. TREM2 is associated with increased risk for Alzheimer’s disease in African Americans. Mol. Neurodegener. 2015, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Baker, M.; Nicholson, A.M.; Rutherford, N.J.; Finch, N.; Soto-Ortolaza, A.; Lash, J.; Wider, C.; Wojtas, A.; DeJesus-Hernandez, M.; et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat. Genet. 2011, 44, 200–205. [Google Scholar] [CrossRef]
- Gerrish, A.; Russo, G.; Richards, A.; Moskvina, V.; Ivanov, D.; Harold, D.; Sims, R.; Abraham, R.; Hollingworth, P.; Chapman, J.; et al. The role of variation at AbetaPP, PSEN1, PSEN2, and MAPT in late onset Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 28, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Cruchaga, C.; Haller, G.; Chakraverty, S.; Mayo, K.; Vallania, F.L.; Mitra, R.D.; Faber, K.; Williamson, J.; Bird, T.; Diaz-Arrastia, R.; et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PLoS ONE 2012, 7, e31039. [Google Scholar] [CrossRef]
- Palmer, M.S.; Dryden, A.J.; Hughes, J.T.; Collinge, J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 1991, 352, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Giau, V.V.; Youn, Y.C.; An, S.S.A.; Kim, S. Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2018, 14, 2067–2085. [Google Scholar] [CrossRef] [PubMed]
- Mead, S.; Uphill, J.; Beck, J.; Poulter, M.; Campbell, T.; Lowe, J.; Adamson, G.; Hummerich, H.; Klopp, N.; Ruckert, I.M.; et al. Genome-wide association study in multiple human prion diseases suggests genetic risk factors additional to PRNP. Hum. Mol. Genet. 2012, 21, 1897–1906. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Wallon, D.; Goupil, C.; Richard, A.-C.; Pottier, C.; Dorval, V.; Sarov-Rivière, M.; Riant, F.; Hervé, D.; Amouyel, P.; et al. Mutation in the 3′untranslated region of APP as a genetic determinant of cerebral amyloid angiopathy. Eur. J. Hum. Genet. 2016, 24, 92–98. [Google Scholar] [CrossRef]
- Murrell, J.; Farlow, M.; Ghetti, B.; Benson, M.D. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science 1991, 254, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Crawford, F.; Houlden, H.; Warren, A.; Hughes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J.; et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 1991, 353, 844–846. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Deming, Y.; Del-Aguila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933. [Google Scholar] [Green Version]
- Marcon, G.; Di Fede, G.; Giaccone, G.; Rossi, G.; Giovagnoli, A.R.; Maccagnano, E.; Tagliavini, F. A novel Italian presenilin 2 gene mutation with prevalent behavioral phenotype. J. Alzheimer’s Dis. 2009, 16, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Schulte, E.C.; Fukumori, A.; Mollenhauer, B.; Hor, H.; Arzberger, T.; Perneczky, R.; Kurz, A.; Diehl-Schmid, J.; Hull, M.; Lichtner, P.; et al. Rare variants in beta-Amyloid precursor protein (APP) and Parkinson’s disease. Eur. J. Hum. Genet. 2015, 23, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A.; Lantos, P.L.; Cairns, N.J. Overlap between neurodegenerative disorders. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2005, 25, 111–124. [Google Scholar] [CrossRef]
- Fernandez, M.V.; Kim, J.H.; Budde, J.P.; Black, K.; Medvedeva, A.; Saef, B.; Deming, Y.; Del-Aguila, J.; Ibanez, L.; Dube, U.; et al. Analysis of neurodegenerative Mendelian genes in clinically diagnosed Alzheimer Disease. PLoS Genet. 2017, 13, e1007045. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Lee, H.-M.; Van Giau, V.; Koh, S.-B.; Jeong, J.H.; An, S.S.A.; Kim, S. PSEN1 p.Thr116Ile Variant in Two Korean Families with Young Onset Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 2604. [Google Scholar] [CrossRef]
- Youn, Y.C.; Lim, Y.K.; Han, S.-H.; Giau, V.V.; Lee, M.-K.; Park, K.-Y.; Kim, S.; Bagyinszky, E.; An, S.S.A.; Kim, H.R. Apolipoprotein ε7 allele in memory complaints: Insights through protein structure prediction. Clin. Interv. Aging 2017, 12, 1095–1102. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Giau, V.; An, S.S. Emergence of exosomal miRNAs as a diagnostic biomarker for Alzheimer’s disease. J. Neurol. Sci. 2016, 360, 141–152. [Google Scholar] [CrossRef]
- Ferrari, R.; Wang, Y.; Vandrovcova, J.; Guelfi, S.; Witeolar, A.; Karch, C.M.; Schork, A.J.; Fan, C.C.; Brewer, J.B.; Momeni, P.; et al. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J. Neurol. Neurosurg. Psychiatry 2017, 88, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.J.; Yi, S.; Han, J.Y.; Park, S.Y.; Jang, J.W.; Chun, I.K.; Giau, V.V.; Bagyinszky, E.; Lim, K.T.; Kang, S.M.; et al. Analysis of Cerebrospinal Fluid and [11C]PIB PET Biomarkers for Alzheimer’s Disease with Updated Protocols. J. Alzheimer’s Dis. 2016, 52, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Giau, V.V.; Lee, H.; Shim, K.H.; Bagyinszky, E.; An, S.S.A. Genome-editing applications of CRISPR-Cas9 to promote in vitro studies of Alzheimer’s disease. Clin. Interv. Aging 2018, 13, 221–233. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]




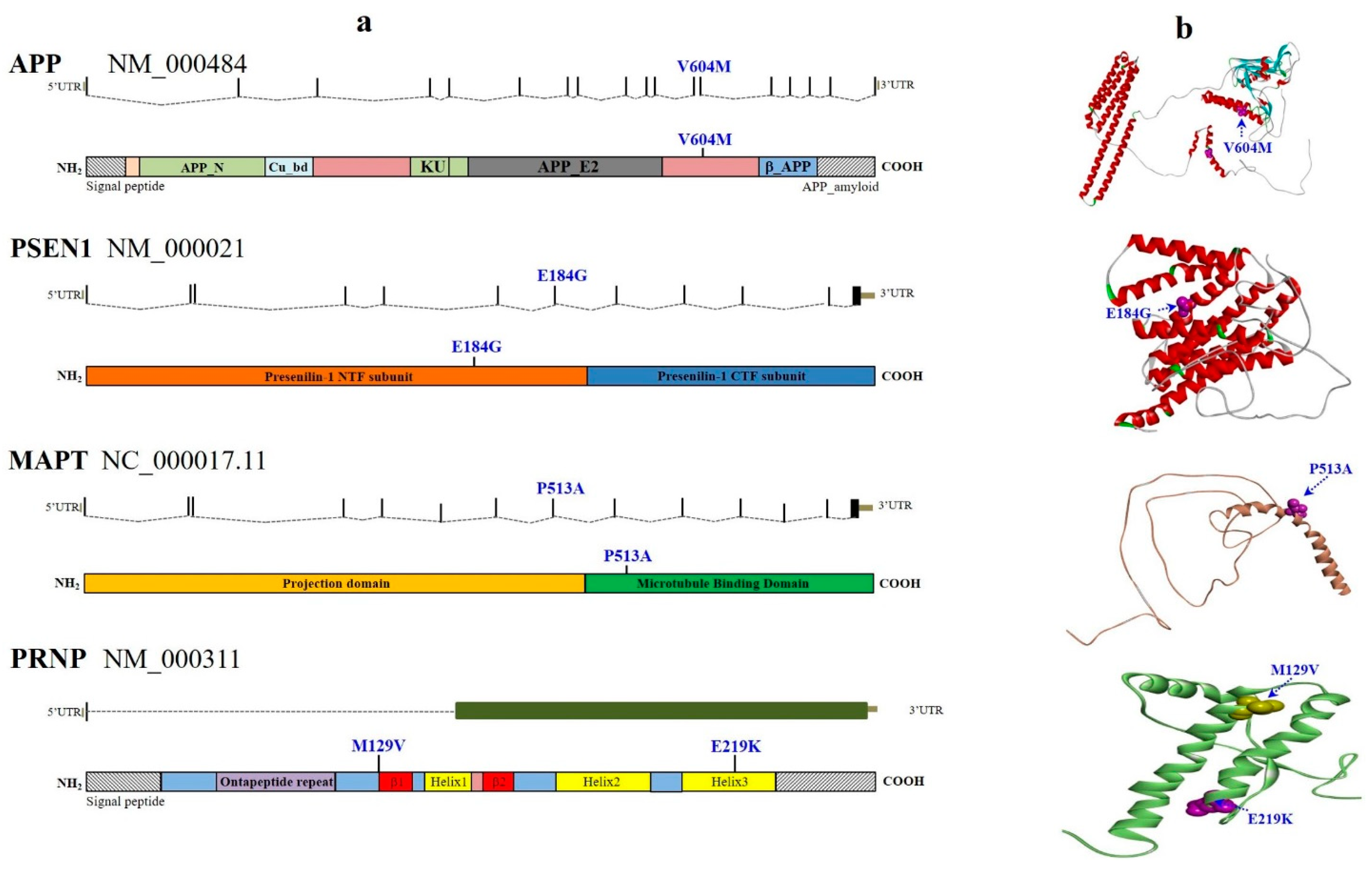{kind=link}
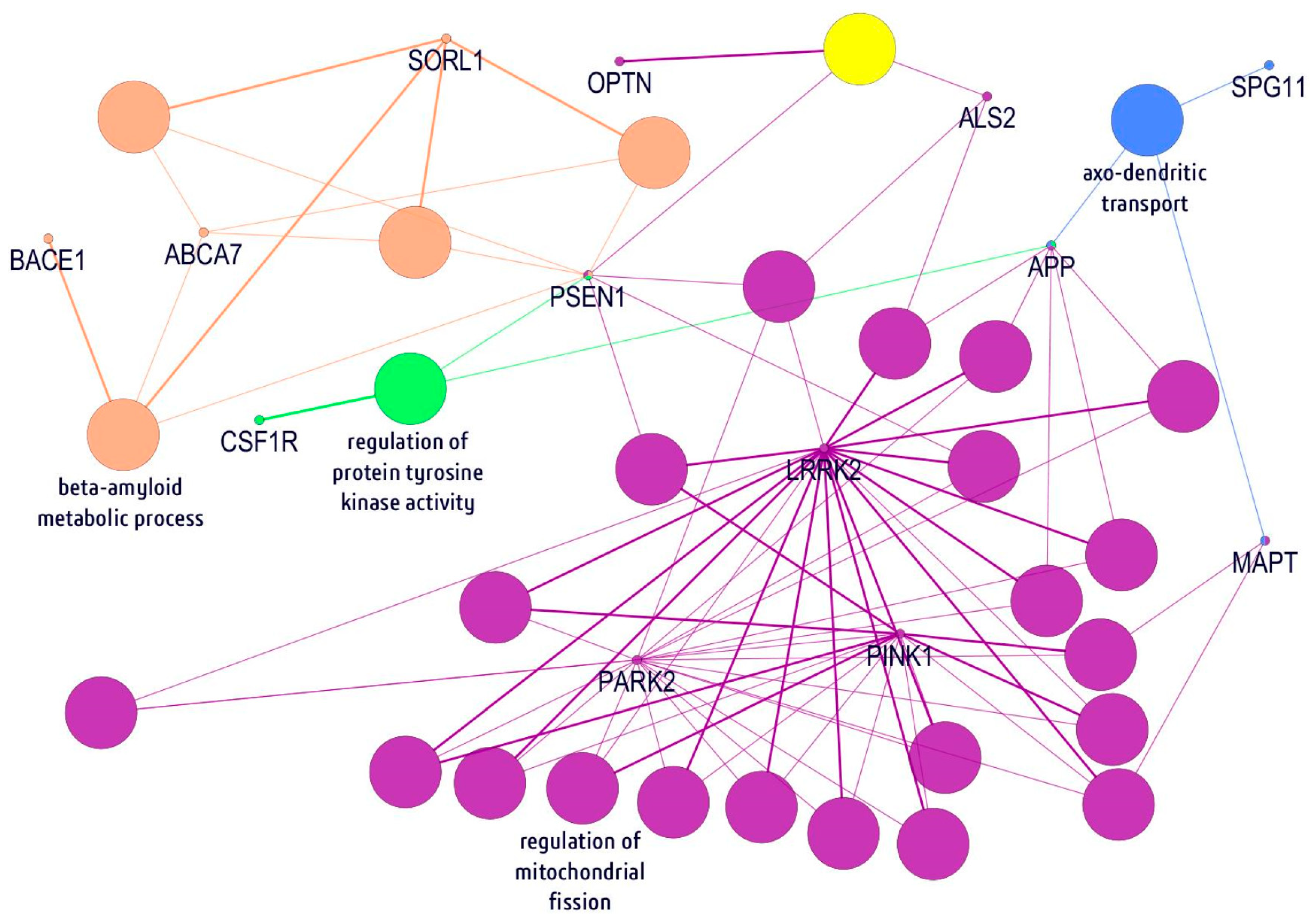{kind=link}
{kind=link}
| Subject No.# | Gender | Age (Years) | APOE | Family History | Clinical Diagnosis | Remark of the Imaging Findings |
|---|---|---|---|---|---|---|
| 1 | Male | 60 | ε3/ε3 | No | AD | Brain single photon emission computed tomography (SPECT) showed decreased uptake at left frontal and left posterior temporal, posterior cingulate gyrus, and most severe at left parietal |
| 2 | Male | 51 | ε3/ε3 | Yes | AD, logophenic aphasia | Magnetic resonance imaging (MRI) showed mild cerebral atrophy with multiple lacunar infraction. Brain SPECT revealed decreased cerebral perfusion at the bilateral temporal, parietal, and the bilateral anterior frontal areas |
| 3 | Male | 56 | ε3/ε3 | Yes | AD | MRI brain showed diffused cerebral atrophy with minimal small vessel disease bilateral at hippocampal atrophy |
| 4 | Female | 51 | ε3/ε3 | No | AD | MRI brain showed diffused cerebral atrophy with bilateral moderate hippocampal atrophy grade IIIEEG study was consistent with moderately severe diffused encephalopathy |
| 5 | Male | 55 | ε3/ε3 | No | AD | MRI brain showed no specific white matter change at periventricular area, whereas SPECT revealed no cerebral perfusion abnormality |
| 6 | Female | 55 | ε3/ε3 | Yes | AD, logophenic aphasia | MRI brain, moderate to severe atrophy of right hippocampusIMP, moderate to severe atrophy of right hippocampus |
| 7 | Female | 41 | ε3/ε3 | Yes | Early-onset dementia | Mini-Mental State Examination (MMSE) was 29/30, Montreal Cognitive Assessment (MoCA) was 25/30 |
| 8 | Female | 60 | ε3/ε4 | No | Early-onset dementia with language impairment | MRI showed mild generalized cerebral cortical atrophy, with a small spot of nonrestrict diffusion. T2/FLAIR analysis revealed hypersignal intensity in the right subcortical frontal lobe |
| Disease Categories | No. of Genes | Candidate Genes Selection |
|---|---|---|
| Alzheimer’s disease | 19 | APP, PSEN1, PSEN2, S100A9, CR1, BIN1, TREM2, CLU, CTNNA3, DNMBP, SORL1, BACE1, PICALM, GAB2, LPR6, ADAM10, ABCA7, CD33, TOMM40. |
| Amyotrophic Lateral Sclerosis (ALS)and Frontotemporal dementia (FTD) | 18 | TDP43, CHMP2B, SIGMAR1, VCP, FUS, GRN, MAPT, UBQLN2, ALS2, TAF15, FIG4, OPTN, DAO, HNRNPA1, SOD1, ANG, VAPB, SQSTM1. |
| Dementia with Lewy Bodies | 7 | PINK1, PARK7, PARK9, GBA, SNCA, PARK2, LRRK2. |
| Other neurodegenerative | 6 | SPAST, CYP7B1, SPG11, CSF1R, NOTCH3, PRNP. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giau, V.V.; Senanarong, V.; Bagyinszky, E.; An, S.S.A.; Kim, S. Analysis of 50 Neurodegenerative Genes in Clinically Diagnosed Early-Onset Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 1514. https://doi.org/10.3390/ijms20061514
Giau VV, Senanarong V, Bagyinszky E, An SSA, Kim S. Analysis of 50 Neurodegenerative Genes in Clinically Diagnosed Early-Onset Alzheimer’s Disease. International Journal of Molecular Sciences. 2019; 20(6):1514. https://doi.org/10.3390/ijms20061514
Chicago/Turabian StyleGiau, Vo Van, Vorapun Senanarong, Eva Bagyinszky, Seong Soo A. An, and SangYun Kim. 2019. "Analysis of 50 Neurodegenerative Genes in Clinically Diagnosed Early-Onset Alzheimer’s Disease" International Journal of Molecular Sciences 20, no. 6: 1514. https://doi.org/10.3390/ijms20061514