The BET Bromodomain Inhibitor I-BET-151 Induces Structural and Functional Alterations of the Heart Mitochondria in Healthy Male Mice and Rats

 , ,
, ,  , , , and
, , , and
Abstract
:
1. Introduction
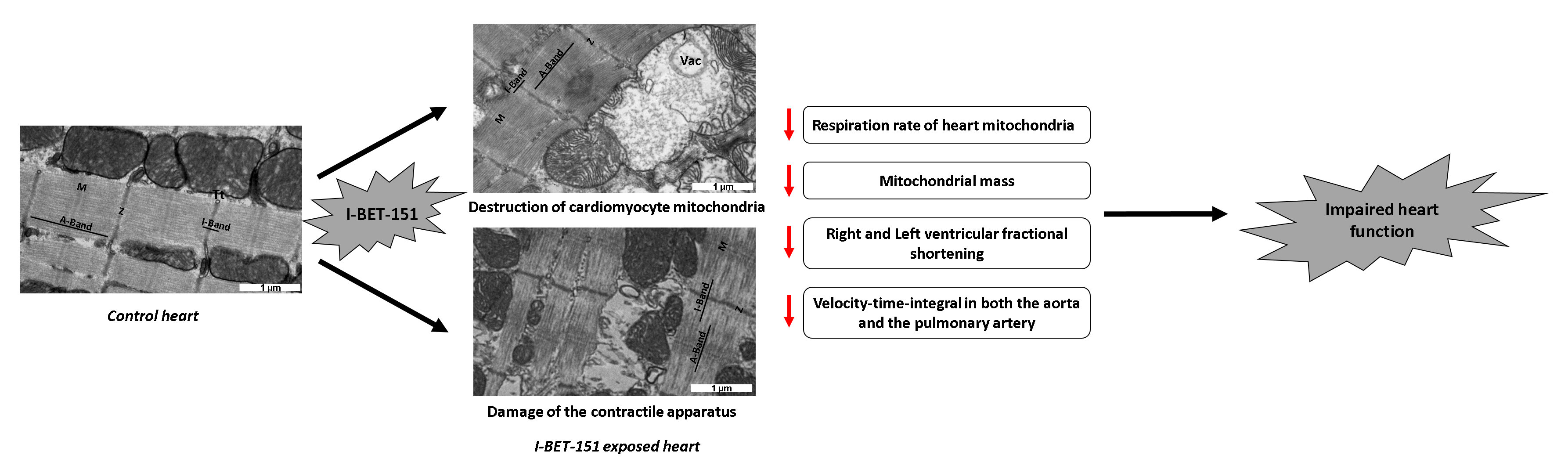
2. Results
2.1. I-BET-151 Induces Heart-Specific Ultrastructural Alterations of Mitochondria in Healthy Male Mice and Rats
2.2. I-BET-151 Decreases Cardiac Mitochondrial Function in Healthy Rats
2.3. I-BET-151-Induced Ultrastructural and Metabolic Alterations of Cardiac Mitochondria Affect Heart Function
3. Discussion
4. Materials and Methods
4.1. In Vivo Studies
4.2. Echocardiographic Evaluation
- -
- In parasternal short axis view: pulmonary artery acceleration time (PAAT), right ventricular ejection time (RVET), cycle length (CL) with heart rate (HR), shape of pulmonary artery outflow pattern, pulmonary artery velocity time integral (VTI Pa), aorta velocity time integral (VTI Aorta), and aorta and Pa valves smallest diameters. Stroke volume (SV) and cardiac output (CO) of left (SV /CO Aorta) and right (SV/CO Pa) outlet tractus were calculated using classical formulas. SV (mL) = VTI × Vessel (Aorta or Pa) surface (cm), and CO (mL/min) = SV × HR. SV and CO are indexed (Si/Ci aorta and Pa) on weight.
- -
- In five cavities view VTI Aorta is analyzed using color and PW dopplers in aorta outlet tractus.
- -
- In four cavities view: RV and LV wall thickness (RVWT, LVWT), RV and LV end-diastolic diameter (RV/LVEDD) and end-systolic diameter (RV/LVESD), interventricular septum shape, TAPSE (Tricuspid Annular Plane Systolic Excursion) in TM Doppler and S tricuspid wave in tissue Doppler imaging coupled with TM.
4.3. Transmission Electron Microscopy (TEM)
4.4. Stereological Analysis
4.5. Mitochondrial Functional Assays in Permeabilized Cardiac Ventricular Fibers
4.6. Enzyme Activity
4.7. Stereological Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMI | acute myocardial infarction |
| BRD | bromodomain |
| BET | bromodomain and extra-terminal domain |
| BETi | bromodomain and extra-terminal domain inhibitor |
| CS | citrate synthase |
| COX | cytochrome oxidase |
| P-TEFb | positive transcription elongation factor |
| TEM | transmission electron microscopy |
| HF | heart failure |
| RV | right ventricle |
| LV | left ventricle |
| VTI | velocity-time-integral |
| RVFS | right ventricular fractional shortening |
| LVFS | left ventricular fractional shortening |
| TAC | transverse aortic constriction |
References
- Fu, L.; Tian, M.; Li, X.; Li, J.; Huang, J.; Ouyang, L.; Zhang, Y.; Liu, B. Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery. Oncotarget 2015, 6, 5501–5516. [Google Scholar] [CrossRef] [Green Version]
- Henssen, A.; Thor, T.; Odersky, A.; Heukamp, L.; El-Hindy, N.; Beckers, A.; Speleman, F.; Althoff, K.; Schäfers, S.; Schramm, A.; et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 2013, 4, 2080–2095. [Google Scholar] [CrossRef] [Green Version]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seal, J.; Lamotte, Y.; Donche, F.; Bouillot, A.; Mirguet, O.; Gellibert, F.; Nicodeme, E.; Krysa, G.; Kirilovsky, J.; Beinke, S.; et al. Identification of a novel series of BET family bromodomain inhibitors: Binding mode and profile of I-BET151 (GSK1210151A). Bioorg. Med. Chem. Lett. 2012, 22, 2968–2972. [Google Scholar] [CrossRef] [PubMed]
- Wyce, A.; Degenhardt, Y.; Bai, Y.; Le, B.; Korenchuk, S.; Crouthame, M.-C.; McHugh, C.F.; Vessella, R.; Creasy, C.L.; Tummino, P.J.; et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget 2013, 4, 2419–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loosveld, M.; Castellano, R.; Gon, S.; Goubard, A.; Crouzet, T.; Pouyet, L.; Prebet, T.; Vey, N.; Nadel, B.; Collette, Y.; et al. Therapeutic targeting of c-Myc in T-cell acute lymphoblastic leukemia, T-ALL. Oncotarget 2014, 5, 3168–3172. [Google Scholar] [CrossRef]
- Qiu, H.; Jackson, A.L.; Kilgore, J.E.; Zhong, Y.; Chan, L.L.-Y.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. JQ1 suppresses tumor growth through downregulating LDHA in ovarian cancer. Oncotarget 2015, 6, 6915–6930. [Google Scholar] [CrossRef]
- Siegel, M.B.; Liu, S.Q.; Davare, M.A.; Spurgeon, S.E.; Loriaux, M.M.; Druker, B.J.; Scott, E.C.; Tyner, J.W. Small molecule inhibitor screen identifies synergistic activity of the bromodomain inhibitor CPI203 and bortezomib in drug resistant myeloma. Oncotarget 2015, 6, 18921–18932. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, A.; Cullinane, C.; De Paoli-Iseppi, R.; Wilmott, J.S.; Gunatilake, D.; Madore, J.; Strbenac, D.; Yang, J.Y.; Gowrishankar, K.; Tiffen, J.C.; et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget 2015, 6, 21507–21521. [Google Scholar] [CrossRef] [Green Version]
- Ambrosini, G.; Sawle, A.D.; Musi, E.; Schwartz, G.K. BRD4-targeted therapy induces Myc-independent cytotoxicity in Gnaq/11-mutatant uveal melanoma cells. Oncotarget 2015, 6, 33397–33409. [Google Scholar] [CrossRef] [Green Version]
- Borbely, G.; Haldosen, L.-A.; Dahlman-Wright, K.; Zhao, C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget 2015, 6, 33623–33635. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-Q.; Guo, W.-Z.; Zhang, Y.; Seng, J.-J.; Zhang, H.-P.; Ma, X.-X.; Zhang, G.; Li, J.; Yan, B.; Tang, H.-W.; et al. Suppression of BRD4 inhibits human hepatocellular carcinoma by repressing MYC and enhancing BIM expression. Oncotarget 2016, 7, 2462–2474. [Google Scholar] [CrossRef] [PubMed]
- Hensel, T.; Giorgi, C.; Schmidt, O.; Calzada-Wack, J.; Neff, F.; Buch, T.; Niggli, F.K.; Schäfer, B.W.; Burdach, S.; Richter, G.H.S. Targeting the EWS-ETS transcriptional program by BET bromodomain inhibition in Ewing sarcoma. Oncotarget 2016, 7, 1451–1463. [Google Scholar] [CrossRef]
- Jacques, C.; Lamoureux, F.; Baud’huin, M.; Rodriguez Calleja, L.; Quillard, T.; Amiaud, J.; Tirode, F.; Rédini, F.; Bradner, J.E.; Heymann, D.; et al. Targeting the epigenetic readers in Ewing sarcoma inhibits the oncogenic transcription factor EWS/Fli1. Oncotarget 2016, 7, 24125–24140. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, R.C.; Clark, P.G.K.; Howarth, A.; Wan, X.; Ceroni, A.; Siejka, P.; Nunez-Alonso, G.A.; Monteiro, O.; Rogers, C.; Gamble, V.; et al. BET inhibition as a new strategy for the treatment of gastric cancer. Oncotarget 2016, 7, 43997–44012. [Google Scholar] [CrossRef] [Green Version]
- French, C.A. Small-Molecule Targeting of BET Proteins in Cancer. Adv. Cancer Res. 2016, 131, 21–58. [Google Scholar]
- Moudgil, R.; Yeh, E.T.H. Mechanisms of Cardiotoxicity of Cancer Chemotherapeutic Agents: Cardiomyopathy and Beyond. Can. J. Cardiol. 2016, 32, 863–870.e5. [Google Scholar] [CrossRef] [PubMed]
- Zamorano, J.L.; Lancellotti, P.; Rodriguez Muñoz, D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef] [PubMed]
- Kerkelä, R.; Grazette, L.; Yacobi, R.; Iliescu, C.; Patten, R.; Beahm, C.; Walters, B.; Shevtsov, S.; Pesant, S.; Clubb, F.J.; et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006, 12, 908–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcieri, E.; Gobbi, P.; Zamai, L.; Vitale, M. Ultrastructural features of apoptosis. Scanning Microsc. 1994, 8, 653–665; discussion 665–666. [Google Scholar] [PubMed]
- Spiltoir, J.I.; Stratton, M.S.; Cavasin, M.A.; Demos-Davies, K.; Reid, B.G.; Qi, J.; Bradner, J.E.; McKinsey, T.A. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2013, 63, 175–179. [Google Scholar] [CrossRef]
- Anand, P.; Brown, J.D.; Lin, C.Y.; Qi, J.; Zhang, R.; Artero, P.C.; Alaiti, M.A.; Bullard, J.; Alazem, K.; Margulies, K.B.; et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 2013, 154, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; McMahon, S.; Anand, P.; Shah, H.; Thomas, S.; Salunga, H.T.; Huang, Y.; Zhang, R.; Sahadevan, A.; Lemieux, M.E.; et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci. Transl. Med. 2017, 9, eaah5084. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Huang, J.; Song, K. BET protein inhibition mitigates acute myocardial infarction damage in rats via the TLR4/TRAF6/NF-κB pathway. Exp Ther. Med. 2015, 10, 2319–2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zengerle, M.; Chan, K.-H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sharma, L.; Lu, J.; Finch, P.; Fletcher, S.; Prochownik, E.V. Structurally diverse c-Myc inhibitors share a common mechanism of action involving ATP depletion. Oncotarget 2015, 6, 15857–15870. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. Therapeutic targeting of Myc-reprogrammed cancer cell metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 369–374. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.-W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.A.; Wang, Y.; Sims-Lucas, S.; Cherok, E.; Rothermund, K.; Branca, M.F.; Elster, J.; Beer-Stolz, D.; Van Houten, B.; Vockley, J.; et al. Mitochondrial structure, function and dynamics are temporally controlled by c-Myc. PLoS ONE 2012, 7, e37699. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Tasdemir, N.; Dow, L.E.; van Es, J.H.; Wilkinson, J.E.; Zhao, Z.; Clevers, H.; Lowe, S.W. Inducible in vivo silencing of Brd4 identifies potential toxicities of sustained BET protein inhibition. Cell Rep. 2014, 8, 1919–1929. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Moulin, M.; Piquereau, J.; Zurlo, G.; Garnier, A. Sex differences in anthracycline cardiotoxicity. Ital. J. Gend. -Specif. Med. 2016, 2, 47–54. [Google Scholar]
- Moulin, M.; Piquereau, J.; Mateo, P.; Fortin, D.; Rucker-Martin, C.; Gressette, M.; Lefebvre, F.; Gresikova, M.; Solgadi, A.; Veksler, V.; et al. Sexual dimorphism of doxorubicin-mediated cardiotoxicity: Potential role of energy metabolism remodeling. Circ. Heart Fail. 2015, 8, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Moulin, M.; Piquereau, J.; Lemaire, C.; Mericskay, M.; Veksler, V.; Garnier, A. Mitochondria: A central target for sex differences in pathologies. Clin. Sci. 2017, 131, 803–822. [Google Scholar] [CrossRef] [PubMed]
- Richter, G.W.; Solez, K. Transition Metal Toxicity; Elsevier: Amsterdam, The Netherlands, 2013; 206p. [Google Scholar]
- Krack, A.; Sharma, R.; Figulla, H.R.; Anker, S.D. The importance of the gastrointestinal system in the pathogenesis of heart failure. Eur. Heart J. 2005, 26, 2368–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranchoux, B.; Bigorgne, A.; Hautefort, A.; Girerd, B.; Sitbon, O.; Montani, D.; Humbert, M.; Tcherakian, C.; Perros, F. Gut-Lung Connection in Pulmonary Arterial Hypertension. Am. J. Respir. Cell Mol. Biol. 2017, 56, 402–405. [Google Scholar] [CrossRef]
- Dores-Sousa, J.L.; Duarte, J.A.; Seabra, V.; de Bastos, M.L.; Carvalho, F.; Costa, V.M. The age factor for mitoxantrone’s cardiotoxicity: Multiple doses render the adult mouse heart more susceptible to injury. Toxicology 2015, 329, 106–119. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, Y.; Toubai, T.; Oravecz-Wilson, K.; Liu, C.; Mathewson, N.; Wu, J.; Rossi, C.; Cummings, E.; Wu, D.; et al. BET bromodomain inhibition suppresses graft-versus-host disease after allogeneic bone marrow transplantation in mice. Blood 2015, 125, 2724–2728. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Gelato, K.A.; Fernández-Montalván, A.; Siegel, S.; Haendler, B. Targeting BET bromodomains for cancer treatment. Epigenomics 2015, 7, 487–501. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Veksler, V.; Gellerich, F.N.; Saks, V.; Margreiter, R.; Kunz, W.S. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat. Protoc. 2008, 3, 965–976. [Google Scholar] [CrossRef]
- Wharton, D.; Tzagoloff, A. Cytochrome oxidase from beef heart mitochondria. Methods Enzymol. 1967, 10, 245–250. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Alterations | Groups | Statistical Analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| W3-RV | W6-RV | W3-LV | W6-LV | ANOVA | W3-RV | W6-RV | W3-LV | W6-LV | ||
| Anomalous | Control | 5.2 ± 2 | 3.2 ± 2.2 | 5.1 ± 2.7 | 2 ± 1.6 | ¶, §, i | - | - | - | - |
| I-BET 2 mg/kg/day | 25 ± 4.3 | 14.5 ± 6.2 | 11.6 ± 3.8 | 13.1 ± 4 | * | - | - | - | ||
| I-BET 10 mg/kg/day | 30.7 ± 6 | 12.2 ± 3.4 | 31.8 ± 5.2 | 26.8 ± 7.1 | ** | - | **, ¤ | ** | ||
| Vacuolized | Control | 2.1 ± 1 | 1.7 ± 1.3 | 2.1 ± 1.2 | 0 ± 0 | ¶, ¥ | - | - | - | - |
| I-BET 2 mg/kg/day | 17.7 ± 5.3 | 14.3 ± 6.2 | 28.8 ± 9 | 23.4 ± 5.2 | - | - | * | * | ||
| I-BET 10 mg/kg/day | 31 ± 9.9 | 4.8 ± 4 | 29.4 ± 11.1 | 34.1 ± 8.2 | * | - | * | * | ||
| Total (A + V) | Control | 7.2 ± 2.8 | 4.9 ± 2.2 | 7.2 ± 3 | 2 ± 1.6 | ¶, §, i | - | - | - | - |
| I-BET 2 mg/kg/day | 42.8 ± 8.6 | 28.7 ± 10.7 | 40.4 ± 9 | 36.5 ± 7.2 | * | - | * | * | ||
| I-BET 10 mg/kg/day | 61.7 ± 10.9 | 17 ± 4.2 | 61.3 ± 8.4 | 60.9 ± 10.7 | *** | $$ | *** | ***, £ | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piquereau, J.; Boet, A.; Péchoux, C.; Antigny, F.; Lambert, M.; Gressette, M.; Ranchoux, B.; Gambaryan, N.; Domergue, V.; Mumby, S.; et al. The BET Bromodomain Inhibitor I-BET-151 Induces Structural and Functional Alterations of the Heart Mitochondria in Healthy Male Mice and Rats. Int. J. Mol. Sci. 2019, 20, 1527. https://doi.org/10.3390/ijms20071527
Piquereau J, Boet A, Péchoux C, Antigny F, Lambert M, Gressette M, Ranchoux B, Gambaryan N, Domergue V, Mumby S, et al. The BET Bromodomain Inhibitor I-BET-151 Induces Structural and Functional Alterations of the Heart Mitochondria in Healthy Male Mice and Rats. International Journal of Molecular Sciences. 2019; 20(7):1527. https://doi.org/10.3390/ijms20071527
Chicago/Turabian StylePiquereau, Jérôme, Angèle Boet, Christine Péchoux, Fabrice Antigny, Mélanie Lambert, Mélanie Gressette, Benoît Ranchoux, Natalia Gambaryan, Valérie Domergue, Sharon Mumby, and et al. 2019. "The BET Bromodomain Inhibitor I-BET-151 Induces Structural and Functional Alterations of the Heart Mitochondria in Healthy Male Mice and Rats" International Journal of Molecular Sciences 20, no. 7: 1527. https://doi.org/10.3390/ijms20071527