Quantum Chemical and Kinetic Study on Radical/Molecule Formation Mechanism of Pre-Intermediates for PCTA/PT/DT/DFs from 2-Chlorothiophenol and 2-Chlorophenol Precursors

Abstract
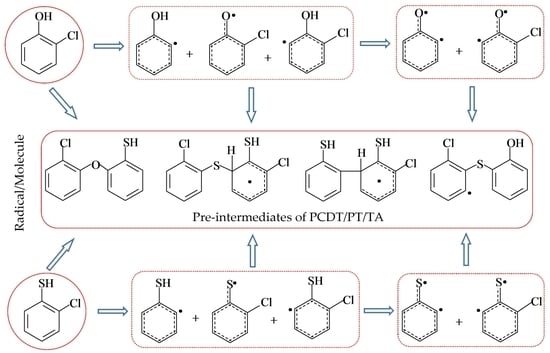
:
1. Introduction
2. Results
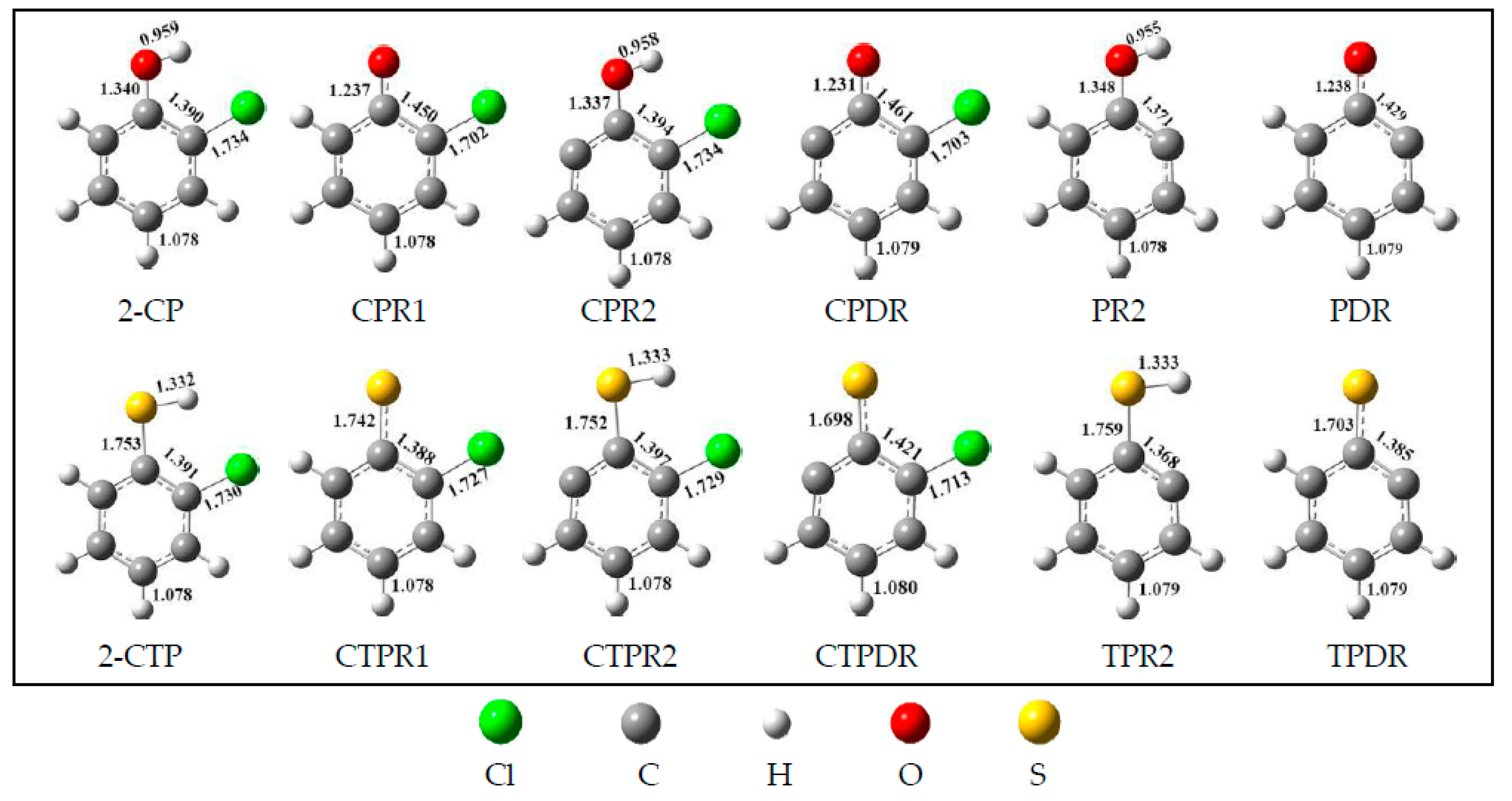
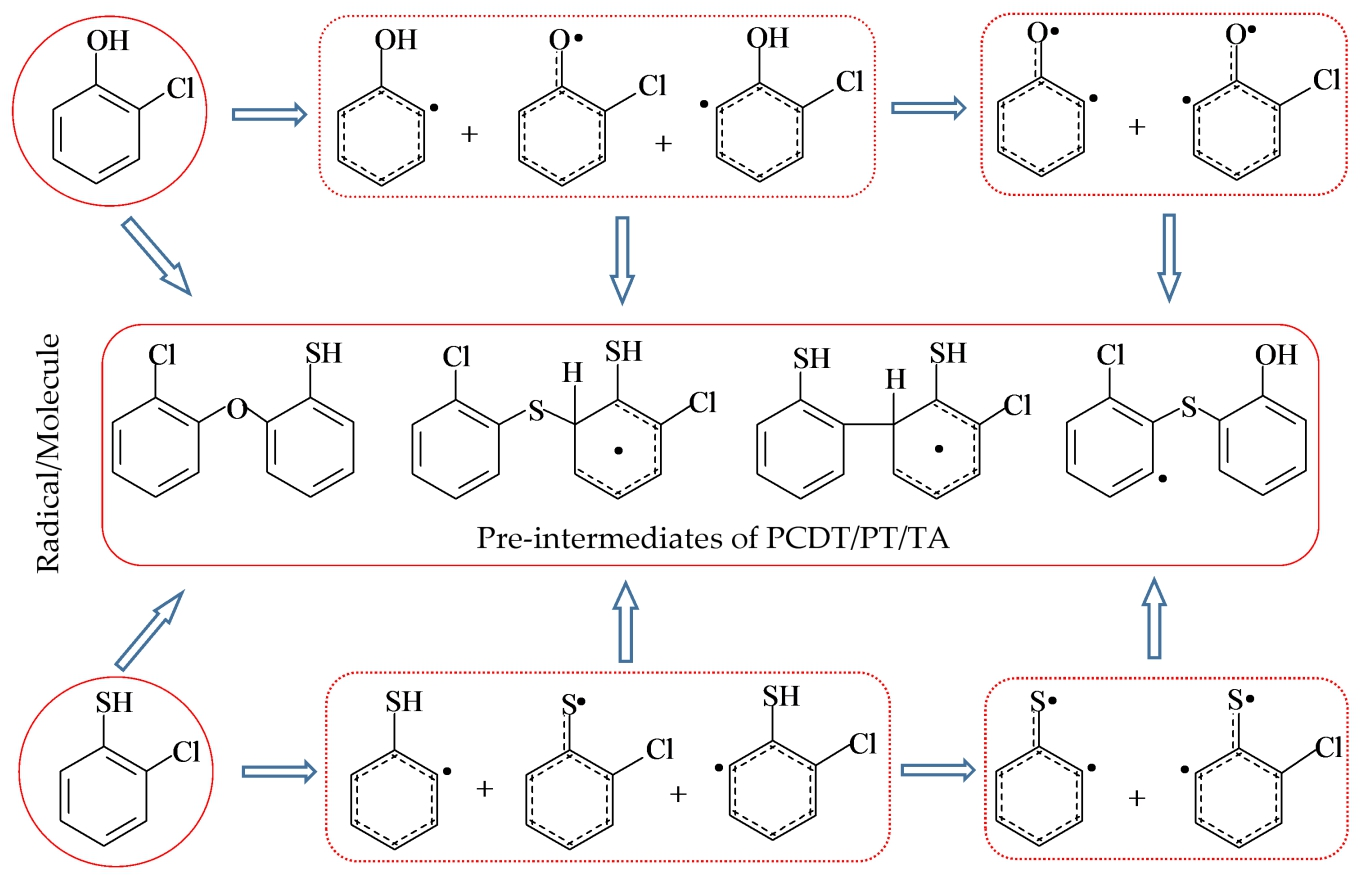
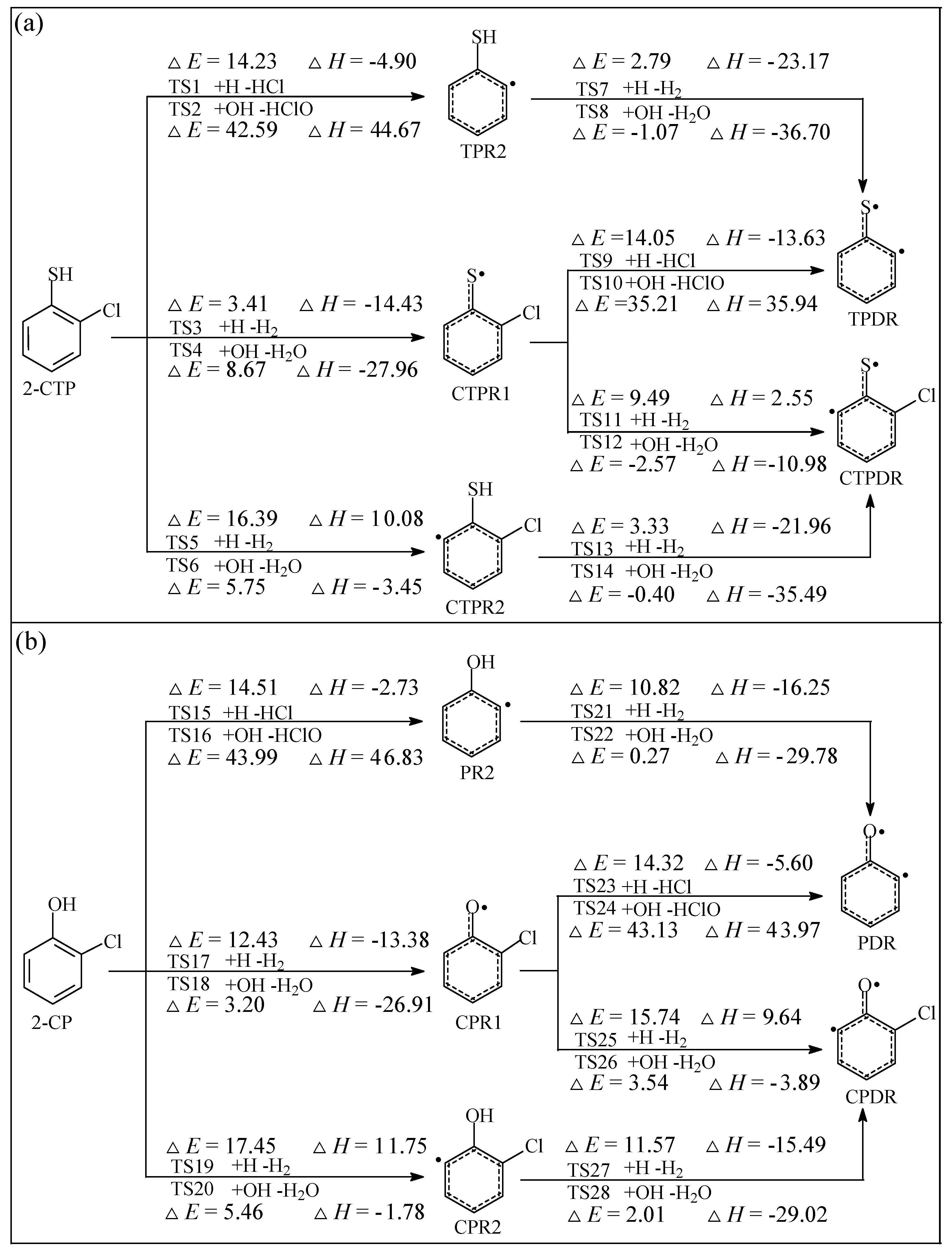
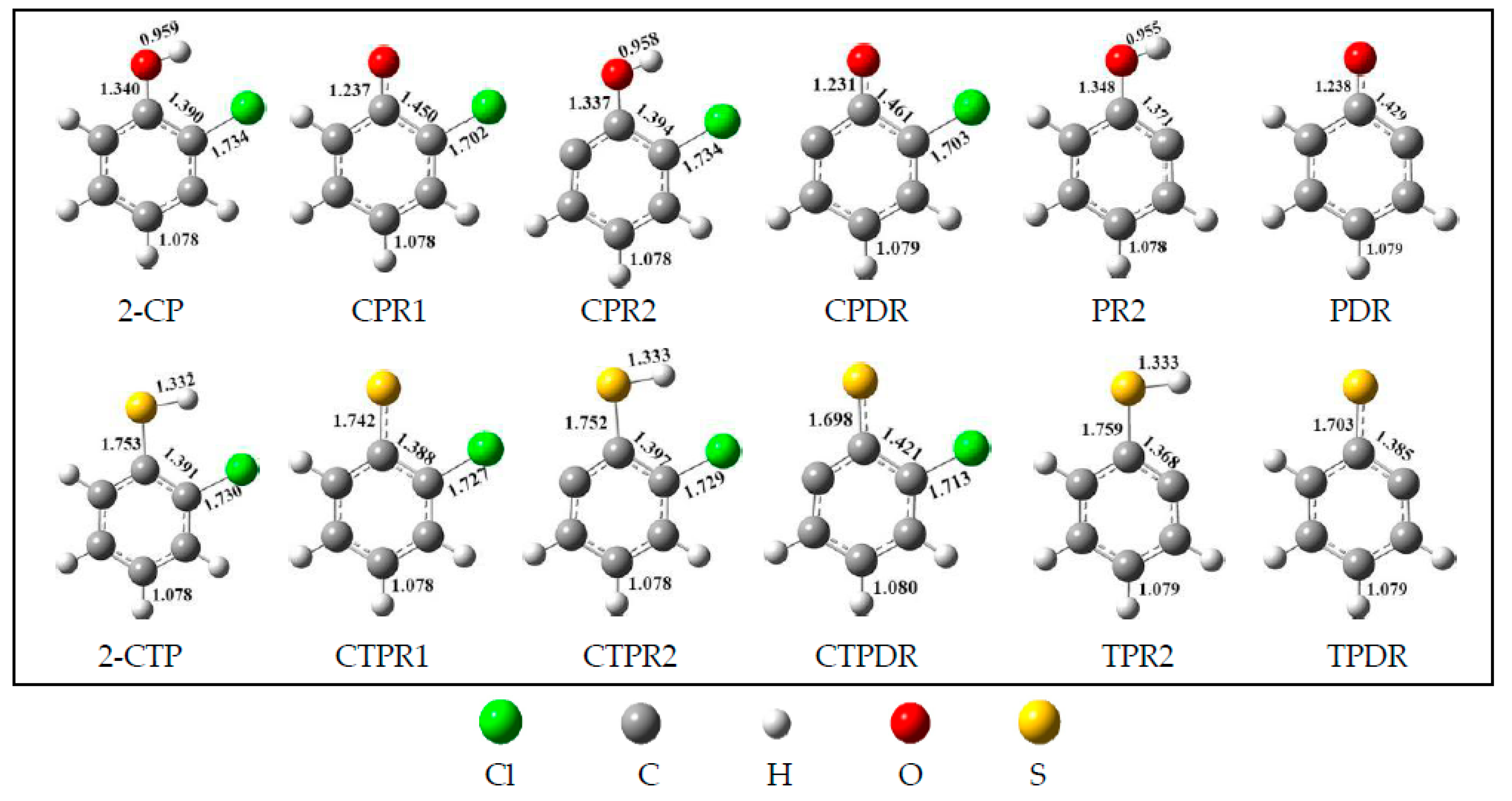
2.1. Formation of Radical Species C(T)PR1, C(T)PR2, C(T)PDR, (T)PR2, and (T)PDR from 2-CTP and 2-CP Molecules
2.2. Formation of Pre-Intermediates of PCTA/PT/DT/DFs via Radical/Molecule Coupling Reactions
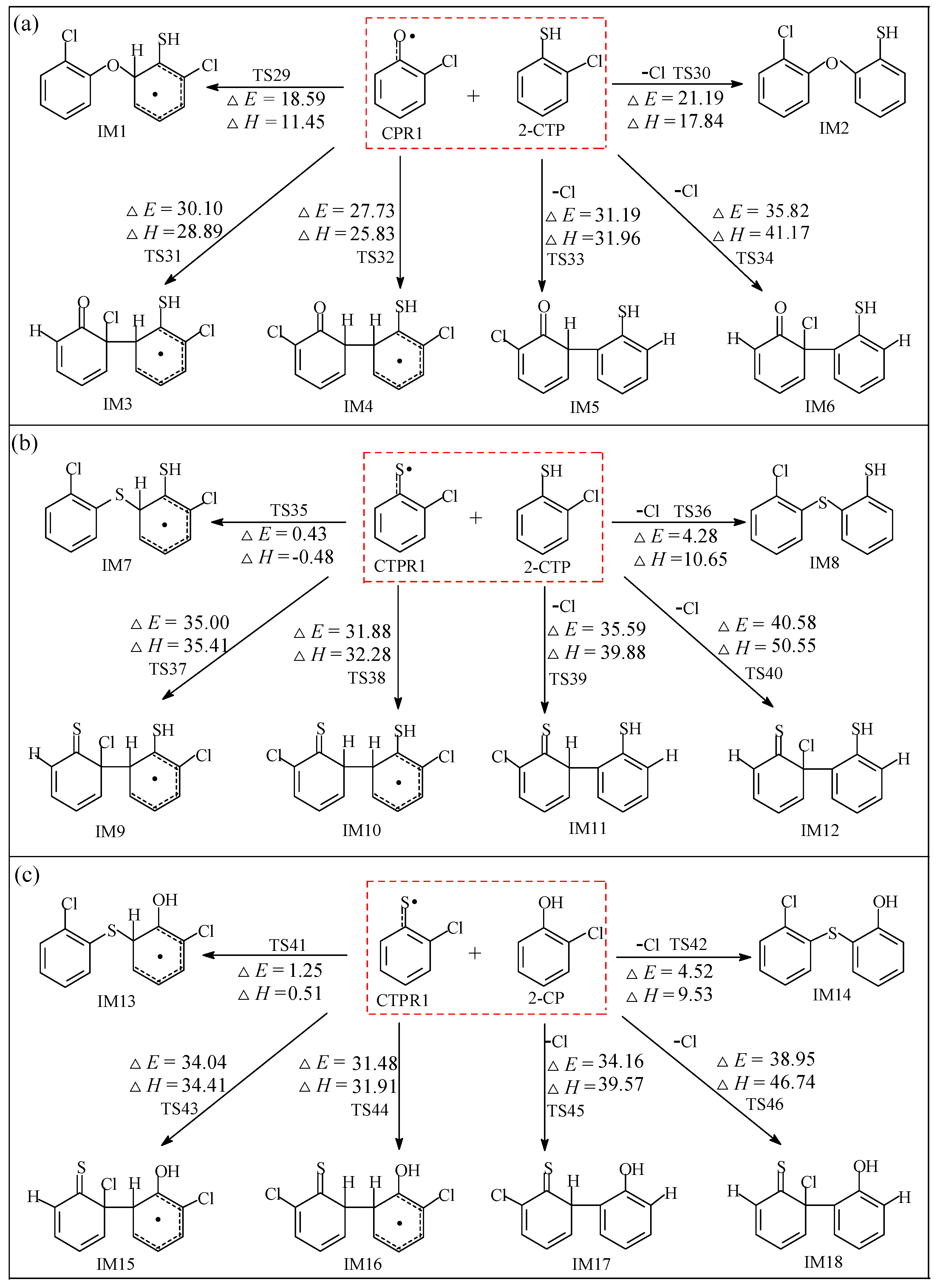
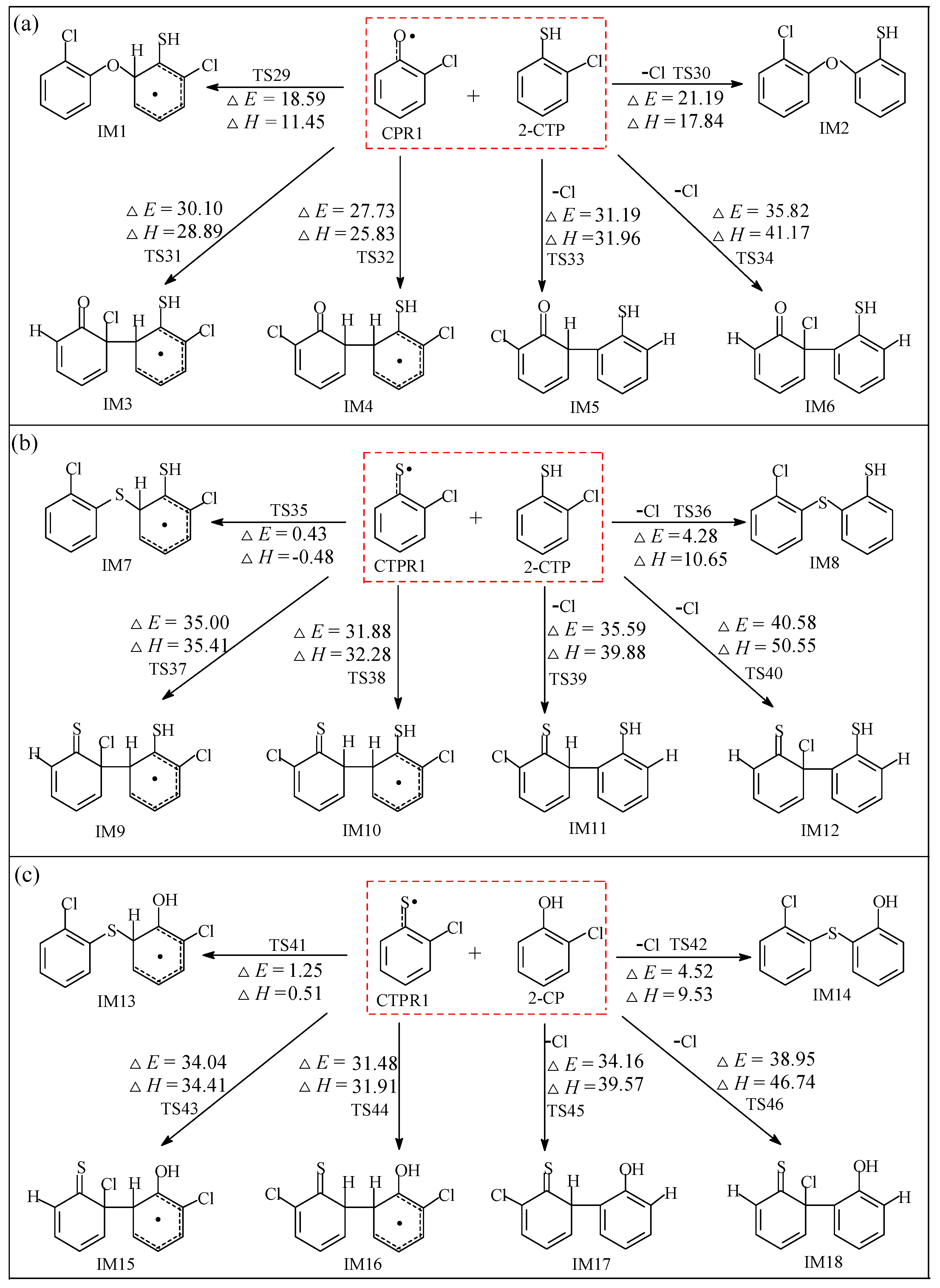
2.2.1. Coupling Reaction of C(T)PR1 with 2-C(T)P
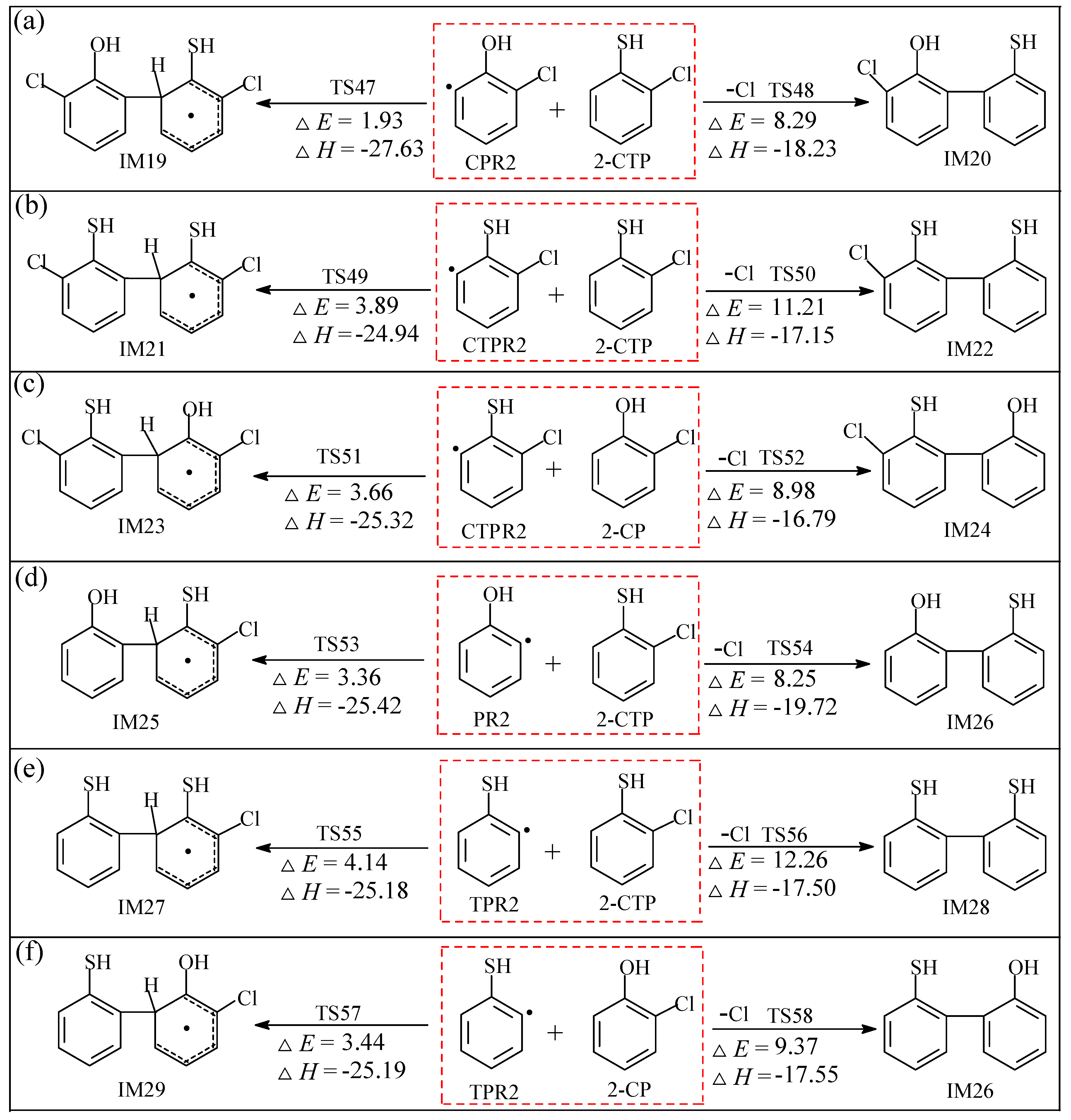
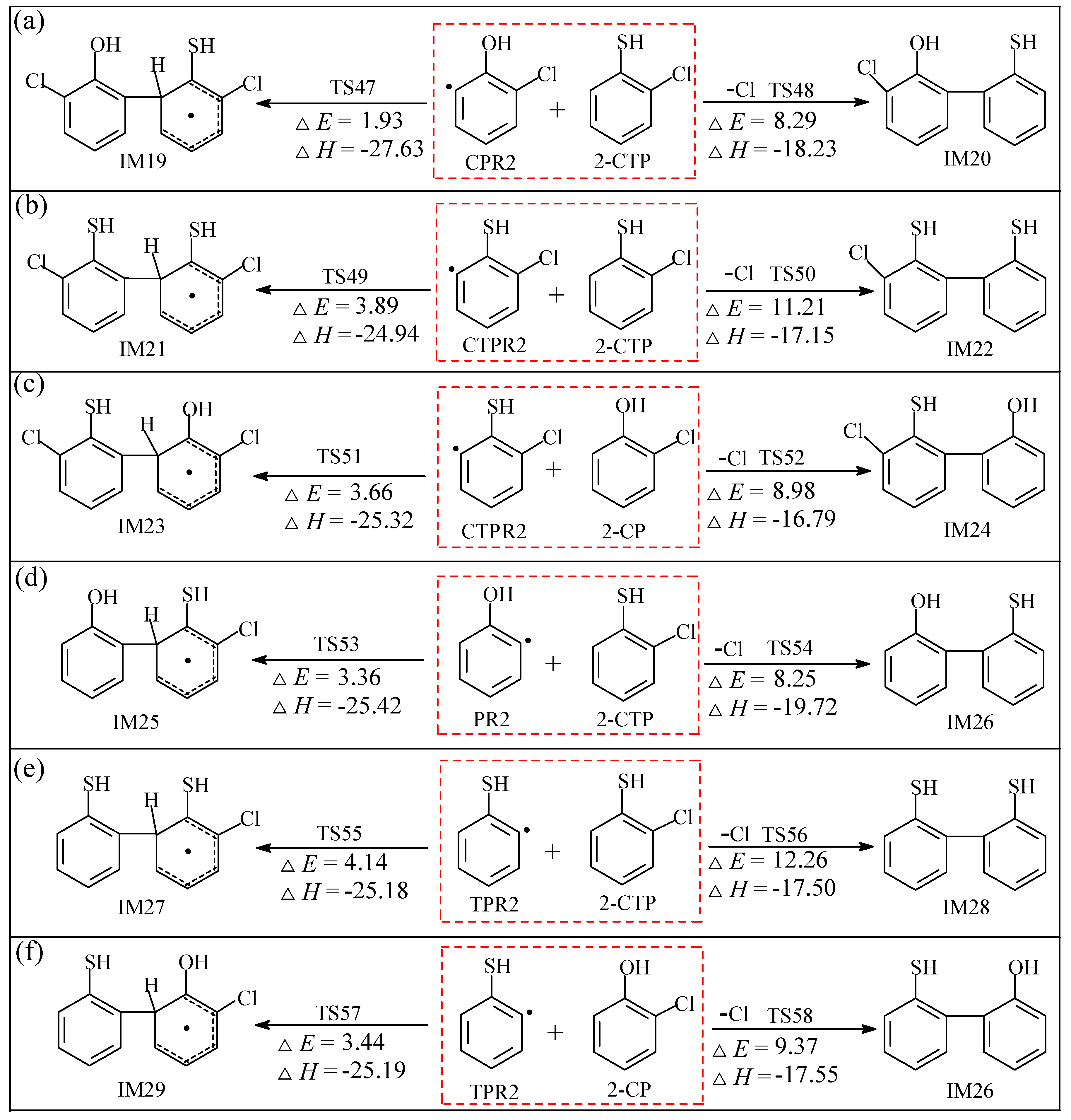
2.2.2. Coupling Reactions of C(T)PR2 and (T)PR2 with 2-C(T)P
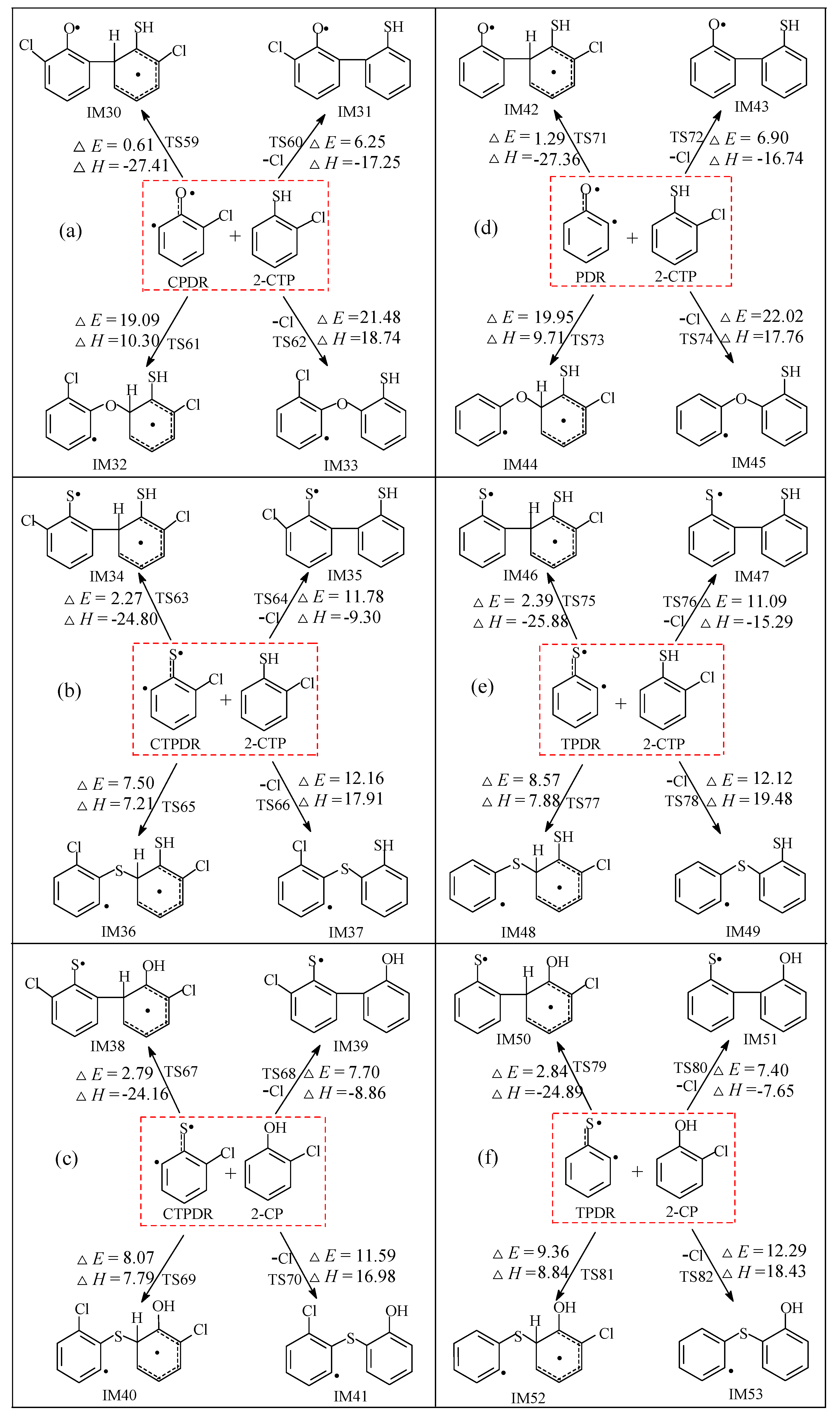
2.2.3. Coupling Reactions of C(T)PDR and (T)PDR with 2-C(T)P
2.3. Rate Constant Calculations
3. Discussion
3.1. Formation of Radical Species C(T)PR1, C(T)PR2, C(T)PDR, (T)PR2, and (T)PDR from 2-CTP and 2-CP Molecules
3.2. Formation of Pre-Intermediates of PCTA/PT/DT/DFs via Radical/Molecule Coupling Reactions
3.2.1. Coupling Reaction of C(T)PR1 with 2-C(T)P
3.2.2. Coupling Reactions of C(T)PR2 and (T)PR2 with 2-C(T)P
3.2.3. Coupling Reactions of C(T)PDR and (T)PDR with 2-C(T)P
3.2.4. Comparing the Reactions of Thiophenoxy Radicals with 2-C(T)P Couplings, Sulfydryl/Hydroxyl-Substituted Phenyl Radicals with 2-C(T)P Couplings, and (thio)phenoxyl Diradicals with 2-C(T)P Couplings
3.3. Rate Constant Calculations
4. Materials and Methods
4.1. Density Functional Theory
4.2. Kinetic Calculation
4.3. Accuracy Verification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PCDTs | polychlorinated dibenzothiophenes |
| PCPTs | polychlorinated phenoxathiins |
| PCTAs | polychlorinated thianthrenes |
| PCDDs | polychlorinated dibenzo-p-dioxins |
| PCDFs | polychlorinated dibenzofurans |
| 2-CP | 2-chlorophenol |
| 2-CTP | 2-chlorothiophenol |
| CPR1 | 2-chlorophenoxy |
| CPR2 | 2-hydroxyl-3-chloro-phenyl |
| CPDR | chlorinated phenoxyl diradical |
| PR2 | 2-hydroxylphenyl radical |
| PDR | phenoxyl diradical |
| CTPR1 | 2-chlorothiophenoxy |
| CTPR2 | 2-sulfydryl-3-chloro-phenyl |
| CTPDR | chlorinated thiophenoxyl diradical |
| TPR2 | 2-sulfydrylphenyl radical |
| TPDR | thiophenoxyl diradical |
| TST | transition state theory |
| IRC | intrinsic reaction coordinate |
| KiSThelP | kinetic and statistical thermodynamic |
| ZPE | zero-point energy |
References
- Wang, Y.; Zeng, X.L.; Chen, H.J.; Wang, H.J. Thermodynamic properties and relative stability of polychlorinated thianthrenes by density functional theory. J. Chem. Eng. Data 2007, 52, 1442–1448. [Google Scholar] [CrossRef]
- Chen, S.D.; Liu, H.X.; Wang, Z.Y. Study of structural and thermodynamic properties for polychlorinated dibenzothiophenes by density functional theory. J. Chem. Eng. Data 2007, 52, 1195–1202. [Google Scholar] [CrossRef]
- Sinkkonen, S. New Types of Persistent Halogenated Compounds; Springer: Berlin, Germany, 2000; Volume 3, pp. 289–314. [Google Scholar]
- Puzyn, T.; Rostkowski, P.; Swieczkowski, A.; Jędrusiak, A.; Falandysz, J. Prediction of environmental partition coefficients and the Henry’s law constants for 135 congeners of chlorodibenzothiophene. Chemosphere 2006, 62, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Kopponen, P.; Sinkkonen, S.; Poso, A.; Gynther, J.; Karenlampi, S. Sulfur analogues of polychlorinated dibenzo-p-dioxins, dibenzofurans and diphenyl ethers as inducers of CYP1A1 in mouse hepatoma cell culture and structure-activity relationships. Environ. Toxicol. Chem. 1994, 13, 1543–1548. [Google Scholar] [CrossRef]
- Weber, R.; Hagenmaier, H.; Schrenk, D. Elimination kinetics and toxicity of 2,3,7,8-tetrachlorothiantheren, a thio analogue of 2,3,7,8-TCDD. Chemosphere 1998, 36, 2635–2641. [Google Scholar] [CrossRef]
- Nakai, S.; Espino, M.P.; Nomura, Y.; Hosomi, M. Detection of polychlorinated dibenzothiophenes (PCDTs) in the environmental samples and investigation of their photodegradability and dioxin-like endocrine disruption potentcy. J. Environ. Chem. 2004, 14, 835–844. [Google Scholar] [CrossRef]
- Buser, H.R.; Rappe, C. Determination of polychlorodibenzothiophenes, the sulfur analogs of polychlorodibenzofurans, using various gas chromatographic/mass spectrometric techniques. Anal. Chem. 1991, 63, 1210–1217. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Paasivirta, J.; Lahtipera, M. Chlorinated and methylated dibenzothiophenes in sediment samples from a river contaminated by organochlorine wastes. J. Soils Sediments 2001, 1, 9–14. [Google Scholar] [CrossRef]
- Pruell, R.J.; Rubinstein, N.I.; Taplin, B.K.; Livolsi, J.A.; Bowen, R.D. Accumulation of polychlorinated organic contaminants from sediment by three benthic marine species. Arch. Environ. Contam. Toxicol. 1993, 24, 290–297. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Kolehmainen, E.; Paasivirta, J.; Koistinen, J.; Lahtipera, M.; Lammi, R. Identification and level estimation of chlorinated neutral aromatic sulfur compounds and their alkylated derivatives in pulp mill effluents and sediments. Chemosphere 1994, 28, 2049–2066. [Google Scholar] [CrossRef]
- Sato, S.; Matsumura, A.; Urushigawa, Y.; Metwally, M.; Al-Muzaini, S. Structural analysis of weathered oil from Kuwait’s environment. Environ. Int. 1998, 24, 77–87. [Google Scholar] [CrossRef]
- Atlas, R.M. Fate of oil from two major oil spills: Role of microbial degradation in removing oil from the Amoco Cadiz and IXTOC I spills. Environ. Int. 1981, 5, 33–38. [Google Scholar] [CrossRef]
- Buser, H.R. Identification and sources of dioxin-like compounds: I. polychlorodibenzothiophenes and polychlorothianthrenes, the sulfur-analogues of the polychlorodibenzofurans and polychlorodibenzodioxins. Chemosphere 1992, 25, 45–48. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Paasivirta, J.; Koistinen, J.; Tarhanen, J. Tetra- and pentachlorodibenzothiophenes are formed in waste combustion. Chemosphere 1991, 23, 583–587. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Kolehmainen, E.; Koistinen, J.; Lahtipera, M. High-resolution gas chromatographic-mass spectrometric determination of neutral chlorinated aromatic sulphur compounds in stack gas samples. J. Chromatogr. A 1993, 641, 309–317. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Vattulainen, A.; Aittola, J.P.; Paasivirta, J.; Tarhanen, J.; Lahtipera, M. Metal reclamation produces sulphur analogues of toxic dioxins and furans. Chemosphere 1994, 28, 1279–1288. [Google Scholar] [CrossRef]
- Sinkkonen, S.; Paasivirta, J.; Koistinen, J.; Lahtipera, M.; Lammi, R. Polychlorinated dibenzothlophenes in bleached pulp mill effluents. Chemosphere 1992, 24, 1755–1763. [Google Scholar] [CrossRef]
- Mostrąg, A.; Puzyn, T.; Haranczyk, M. Modeling the overall persistence and environmental mobility of sulfur-containing polychlorinated organic compounds. Environ. Sci. Pollut. Res. 2010, 17, 470–477. [Google Scholar] [CrossRef]
- Wiedmann, T.; Riehle, U.; Kurz, J.; Ballschmiter, K. HRGC-MS of polychlorinated phenanthrenes (PCPhen), dibenzothiophenes (PCDT), dibenzothianthrenes (PCTA), and phenoxathiins (PCPT). Fresenius J. Anal. Chem. 1997, 359, 176–188. [Google Scholar] [CrossRef]
- Ferrario, E. Preparation of phenoxathiin from diphenyl ether and sulfur. Bull. Soc. Chim. 1911, 9, 536–537. [Google Scholar]
- Bourke, J.B.; Felsot, A.S.; Gilding, T.J.; Jensen, J.K.; Seiber, J.N. Pesticide Waste Management; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1992; pp. 157–165. [Google Scholar]
- Dar, T.; Altarawneh, M.; Dlugogorski, B. Theoretical study in the dimerisation of 2-chlrothiophenol/2-chlorothiophenoxy: Precursors to PCDT/TA. Organohalogen Compd. 2012, 74, 657–660. [Google Scholar]
- Dar, T.; Altarawneh, M.; Dlugogorski, B.Z. Quantum chemical study on formation of PCDT/TA from 2-chlorothiophenol precursor. Environ. Sci. Technol. 2013, 47, 11040–11047. [Google Scholar] [CrossRef]
- Dar, T.; Shah, K.; Moghtaderi, B.; Page, A.J. Formation of persistent organic pollutants from 2,4,5-trichlorothiophenol combustion: A density functional theory investigation. J. Mol. Model. 2016, 22, 128. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Shi, X.L.; Li, Y.F.; Zhang, Q.Z. Mechanistic and kinetic studies on the homogeneous gas-phase formation of PCTA/DTs from 2,4-dichlorothiophenol and 2,4,6-trichlorothiophenol. Int. J. Mol. Sci. 2015, 16, 20449–20467. [Google Scholar] [CrossRef]
- Yu, X.Q.; Chang, J.M.; Liu, X.; Pan, W.X.; Zhang, A.Q. Theoretical study on the formation mechanism of polychlorinated dibenzothiophenes/thianthrenes from 2-chlorothiophenol molecules. J. Environ. Sci. 2018, 66, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Parette, R.; Pearson, W.N. 2,4,6,8-Tetrachlorodibenzothiophene in the Newark Bay Estuary: The likely source and reaction pathways. Chemosphere 2014, 111, 157–163. [Google Scholar] [CrossRef]
- Navarro, R.; Bierbrauer, K.; Mijangos, C.; Goiti, E.; Reinecke, H. Modification of poly (vinyl chloride) with new aromatic thiol compounds. Synthesis and characterization. Polym. Degrad. Stab. 2008, 93, 585–591. [Google Scholar] [CrossRef]
- Buisson, R.; Kirk, P.; Lester, J. Determination of chlorinated phenols in water, wastewater, and wastewater sludge by capillary GC/ECD. J. Chromatogr. Sci. 1984, 22, 339–342. [Google Scholar] [CrossRef]
- Wegman, R.C.C.; Van den Broek, H.H. Chlorophenols in river sediment in the Netherlands. Water Res. 1983, 17, 227–230. [Google Scholar] [CrossRef]
- Briois, C.; Visez, N.; Baillet, C.; Sawerysyn, J.P. Experimental study on the thermal oxidation of 2-chlorophenol in air over the temperature range 450–900 °C. Chemosphere 2006, 62, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.; Parker, D.; Zhang, F.; Landera, A.; Kislov, V.; Mebel, A. PAH formation under single collision conditions: Reaction of phenyl radical and 1,3-butadiene to form 1,4-dihydronaphthalene. J. Phys. Chem. A 2012, 116, 4248–4258. [Google Scholar] [CrossRef] [PubMed]
- Shukla, B.; Susa, A.; Miyoshi, A.; Koshi, M. Role of phenyl radicals in the growth of polycyclic aromatic hydrocarbons. J. Phys. Chem. A 2008, 112, 2362–2369. [Google Scholar] [CrossRef] [PubMed]
- Bonnichon, F.; Richard, C.; Grabner, G. Formation of an α-ketocarbene by photolysis of aqueous 2-bromophenol. Chem. Commun. 2001, 73–74. [Google Scholar] [CrossRef]
- Pan, W.X.; Zhang, D.J.; Han, Z.; Zhan, J.H.; Liu, C.B. New insight into the formation mechanism of PCDD/Fs from 2-chlorophenol precursor. Environ. Sci. Technol. 2013, 47, 8489–8498. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.S.; Dellinger, B. Mechanisms of dioxin formation from the high-temperature pyrolysis of 2-chlorophenol. Environ. Sci. Technol. 2003, 37, 1325–1330. [Google Scholar] [CrossRef]
- Evans, C.S.; Dellinger, B. Mechanisms of dioxin formation from the high-temperature oxidation of 2-chlorophenol. Environ. Sci. Technol. 2005, 39, 122–127. [Google Scholar] [CrossRef]
- Zhang, Q.Z.; Li, S.Q.; Qu, X.H.; Shi, X.Y.; Wang, W.X. A quantum mechanical study on the formation of PCDD/Fs from 2-chlorophenol as precursor. Environ. Sci. Technol. 2008, 42, 7301–7308. [Google Scholar] [CrossRef]
- Qu, X.H.; Wang, H.; Zhang, Q.Z.; Shi, X.Y.; Xu, F.; Wang, W.X. Mechanistic and kinetic studies on the homogeneous gas-phase formation of PCDD/Fs from 2,4,5-trichlorophenol. Environ. Sci. Technol. 2009, 43, 4068–4075. [Google Scholar] [CrossRef]
- Pan, W.X.; Fu, J.J.; Zhang, A.Q. Theoretical study on the formation mechanism of pre-intermediates for PXDD/Fs from 2-Bromophenol and 2-Chlorophenol precursors via radical/molecule reactions. Environ. Pollut. 2017, 225, 439–449. [Google Scholar] [CrossRef]
- Sinkkonen, S. Sources and environmental fate of PCDTs. Toxicol. Environ. Chem. 1998, 66, 105–112. [Google Scholar] [CrossRef]
- Czerwinski, J. Pathways of polychlorinated dibenzothiophenes (PCDTs) in the environment. Arch. Environ. Prot. 2008, 34, 169–181. [Google Scholar]
- Zhang, Q.Z.; Qu, X.H.; Wang, H.; Xu, F.; Shi, X.Y.; Wang, W.X. Mechanism and thermal rate constants for the complete series reactions of chlorophenols with H. Environ. Sci. Technol. 2009, 43, 4105–4112. [Google Scholar] [CrossRef]
- Xu, F.; Wang, H.; Zhang, Q.Z.; Zhang, R.X.; Qu, X.H.; Wang, W.X. Kinetic properties for the complete series reactions of chlorophenols with OH radicals–relevance for dioxin formation. Environ. Sci. Technol. 2010, 44, 1399–1404. [Google Scholar] [CrossRef]
- Xu, F.; Shi, X.L.; Zhang, Q.Z.; Wang, W.X. Formation of chlorotriophenoxy radicals from complete series reactions of chlorotriophenols with H and OH radicals. Int. J. Mol. Sci. 2015, 16, 18714–18731. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.S.N.; Kaiser, R.I.; Troy, T.P.; Kostko, O.; Ahmed, M.; Mebel, A.M. Toward the oxidation of the phenyl radical and prevention of PAH formation in combustion systems. J. Phys. Chem. A 2014, 119, 7145–7154. [Google Scholar] [CrossRef] [PubMed]
- Kislov, V.V.; Mebel, A.M. Ab initio/RRKM-ME study on the mechanism and kinetics of the reaction of phenyl radical with 1,2-butadiene. J. Phys. Chem. A 2010, 114, 7682–7692. [Google Scholar] [CrossRef]
- Canneaux, S.; Bohr, F.; Henon, E. KiSThelP: A program to predict thermodynamic properties and rate constants from quantum chemistry results. J. Comput. Chem. 2014, 35, 82–93. [Google Scholar] [CrossRef]
- Wigner, E. Calculation of the rate of elementary association reactions. J. Chem. Phys. 1937, 5, 720–725. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Choi, K.C.; Mulholland, J.A. Polychlorinated dibenzo-p-dioxin (PCDD) and dibenzofurna (PCDF) isomer patterns from municipal waste combustion: Formation mechanism fingerprints. Chemosphere 2006, 65, 1526–1536. [Google Scholar] [CrossRef]
- Altarawneh, M.; Dlugogorski, B.Z.; Kennedy, E.M.; Mackie, J.C. Mechanisms for formation, chlorination, dechlorination and destruction of polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/Fs). Prog. Energy Combust. Sci. 2009, 35, 245–274. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Mulholland, J.A.; Dunn, J.E.; Iino, F.; Gullett, B.K. Potential role of chlorination pathways in PCDD/F formation in a municipal waste incinerator. Environ. Sci. Technol. 2004, 38, 5112–5119. [Google Scholar] [CrossRef]
- Tuppurainen, K.A.; Ruokojarvi, P.H.; Asikainen, A.H.; Aatamila, M.; Ruuskanen, J. Chlorophenols as precursors of PCDD/Fs in incineration processes: Correlations, PLS modeling, and reaction mechanisms. Environ. Sci. Technol. 2000, 34, 4958–4962. [Google Scholar] [CrossRef]
- Shaub, W.M.; Tsang, W. Dioxin formation in incinerators. Environ. Sci. Technol. 1983, 17, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. Hybrid meta density functional theory methods for therochemistry, thermochemical kinetics, and noncovalent interactions: The MPW1B95 and MPWB1K models and comparative assessments for hydrogen bonding and van der Waals interactions. J. Phys. Chem. A 2004, 108, 6908–6918. [Google Scholar] [CrossRef]
- Cao, H.J.; He, M.X.; Sun, Y.H.; Han, D.D. Mechanical and kinetic studies of the formation of polyhalogenated dibenzo-p-dioxins from hydroxylated polybrominated diphenyl ethers and chlorinated derivatives. J. Phys. Chem. A 2011, 115, 13489–13497. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Sun, X.M.; Zhang, C.X.; Gong, C.; Hu, J.T.; Zhang, J.H. Mechanism and kinetics study on the ozonolysis reaction of 2,3,7,8-TCDD in the atmosphere. J. Environ. Sci. 2014, 26, 181–188. [Google Scholar] [CrossRef]
- Zhao, Y.Y.; Sun, X.M.; Bai, J.; Zhang, C.X.; Zhang, J.H. Atmospheric degradation of 2,3,7,8-tetrachlorinated dibenzo-p-dioxins in the presence of NO3 at night. Can. J. Chem. 2012, 91, 398–405. [Google Scholar] [CrossRef]
- Cao, H.J.; He, M.X.; Han, D.D.; Sun, Y.H.; Zhao, S.F.; Ma, H.J.; Yao, S.D. Mechanistic and kinetic study on the reaction of 2;4-dibrominated diphenyl ether (BDE-7) with OH radicals. Comput. Theor. Chem. 2012, 983, 31–37. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Lee, S.Y. Density functional theory calculation of molecular structure and vibrational spectra of dibenzothiophene in the ground and the lowest triplet state. J. Phys. Chem. A 2001, 105, 8093–8097. [Google Scholar] [CrossRef]
- Larson, N.W.; Nicolaisen, F.M. Far-infrared gas spectra of phenol, 4-fluorophenol, thiophenol and some deuterated species: Barrier to internal rotation. J. Mol. Struct. 1974, 22, 29–43. [Google Scholar] [CrossRef]
- Nyquist, R.A.; Evans, J.C. The vibrational spectra of p-chlorobenzenethiol. Spectrochim. Acta 1961, 17, 795–801. [Google Scholar] [CrossRef]
- Johnson, R.D., III. NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database; Department of Commerce: Washington, DC, USA; Release 16a; August 2013. Available online: http://cccbdb.nist.gov/ (accessed on 15 March 2019).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reactions Arrhenius Formulas | Arrhenius Formulas |
|---|---|
| CPR1 + 2-CTP → IM1 via TS29 | k(T) = (2.09 × 10−15) exp (−11163/T) |
| CPR1 + 2-CTP → IM2 + Cl via TS30 | k(T) = (1.79 × 10−15) exp (−12526/T) |
| CPR1 + 2-CTP → IM3 via TS31 | k(T) = (1.43 × 10−15) exp (−17118/T) |
| CPR1 + 2-CTP → IM4 + Cl via TS32 | k(T) = (3.90 × 10−15) exp (−15840/T) |
| CPR1 + 2-CTP → IM5 via TS33 | k(T) = (1.42 × 10−15) exp (−17613/T) |
| CPR1 + 2-CTP → IM6 + Cl TS34 | k(T) = (1.21 × 10−16) exp (−19231/T) |
| CTPR1 + 2-CTP → IM7 via TS35 | k(T) = (2.37 × 10−15) exp (−2233/T) |
| CTPR1 + 2-CTP → IM8 via TS36 | k(T) = (1.22 × 10−15) exp (−4093/T) |
| CTPR1 + 2-CTP → IM9 via TS37 | k(T) = (1.40 × 10−15) exp (−19723/T) |
| CTPR1 + 2-CTP → IM10 via TS38 | k(T) = (5.33 × 10−15) exp (−18080/T) |
| CTPR1 + 2-CTP → IM11 via TS39 | k(T) = (1.99 × 10−15) exp (−19993/T) |
| CTPR1 + 2-CTP → IM12 + Cl TS40 | k(T) = (5.24 × 10−16) exp (−22527/T) |
| CTPR1 + 2-CP → IM13 via TS41 | k(T) = (1.14 × 10−14) exp (−2790/T) |
| CTPR1 + 2-CP → IM14 + Cl via TS42 | k(T) = (1.01 × 10−14) exp (−4524/T) |
| CTPR1 + 2-CP → IM15 via TS43 | k(T) = (6.13 × 10−15) exp (−19327/T) |
| CTPR1 + 2-CP → IM16 + Cl via TS44 | k(T) = (1.06 × 10−14) exp (−17908/T) |
| CTPR1 + 2-CP → IM17 via TS45 | k(T) = (4.46 × 10−15) exp (−19296/T) |
| CTPR1 + 2-CP → IM18 + Cl TS46 | k(T) = (1.29 × 10−15) exp (−21760/T) |
| CPR2 + 2-CTP → IM19 via TS47 | k(T) = (1.42 × 10−14) exp (−3082/T) |
| CPR2 + 2-CTP → IM20 + Cl via TS48 | k(T) = (3.37 × 10−15) exp (−6141/T) |
| CTPR2 + 2-CTP → IM21 via TS49 | k(T) = (7.46 × 10−15) exp (−3994/T) |
| CTPR2 + 2-CTP → IM22 + Cl via TS50 | k(T) = (1.20 × 10−15) exp (−7520/T) |
| CTPR2 + 2-CP → IM23 via TS51 | k(T) = (3.19 × 10−14) exp (−4022/T) |
| CTPR2 + 2-CP → IM24 + Cl TS52 | k(T) = (5.48 × 10−15) exp (−6518/T) |
| PR2 + 2-CTP → IM25 via TS53 | k(T) = (1.26 × 10−14) exp (−3668/T) |
| PR2 + 2-CTP → IM26 + Cl via TS54 | k(T) = (1.53 × 10−15) exp (−5987/T) |
| TPR2 + 2-CTP → IM27 via TS55 | k(T) = (1.21 × 10−14) exp (−4148/T) |
| TPR2 + 2-CTP → IM28 + Cl via TS56 | k(T) = (1.21 × 10−15) exp (−7993/T) |
| TPR2 + 2-CP → IM29 via TS57 | k(T) = (4.85 × 10−14) exp (−3879/T) |
| TPR2 + 2-CP → IM26 + Cl via TS58 | k(T) = (9.06 × 10−15) exp (−6757/T) |
| Reactions Arrhenius Formulas | Arrhenius Formulas |
|---|---|
| CPDR + 2-CTP → IM30 via TS59 | k(T) = (3.33 × 10−14) exp (−2430/T) |
| CPDR + 2-CTP → IM31 + Cl via TS60 | k(T) = (8.91 × 10−15) exp (−5222/T) |
| CPDR + 2-CTP → IM32 via TS61 | k(T) = (2.36 × 10−15) exp (−11424/T) |
| CPDR + 2-CTP → IM33 + Cl via TS62 | k(T) = (5.26 × 10−15) exp (−12811/T) |
| CTPDR + 2-CTP → IM34 via TS63 | k(T) = (1.05 × 10−14) exp (−3225/T) |
| CTPDR + 2-CTP → IM35 + Cl via TS64 | k(T) = (2.45 × 10−15) exp (−7902/T) |
| CTPDR + 2-CTP → IM36 via TS65 | k(T) = (5.65 × 10−15) exp (−5904/T) |
| CTPDR + 2-CTP → IM37 + Cl via TS66 | k(T) = (2.03 × 10−15) exp (−8446/T) |
| CTPDR + 2-CP → IM38 via TS67 | k(T) = (1.43 × 10−13) exp (−3672/T) |
| CTPDR + 2-CP → IM39 + Cl via TS68 | k(T) = (1.72 × 10−14) exp (−6042/T) |
| CTPDR + 2-CP → IM40 via TS69 | k(T) = (1.31 × 10−14) exp (−6256/T) |
| CTPDR + 2-CP → IM41 + Cl TS70 | k(T) = (1.04 × 10−14) exp (−8632/T) |
| PDR + 2-CTP → IM42 via TS71 | k(T) = (3.51 × 10−14) exp (−2775/T) |
| PDR + 2-CTP → IM43 + Cl via TS72 | k(T) = (4.24 × 10−15) exp (−5424/T) |
| PDR + 2-CTP → IM44 via TS73 | k(T) = (6.46 × 10−15) exp (−11909/T) |
| PDR + 2-CTP → IM45 + Cl TS74 | k(T) = (2.54 × 10−14) exp (−13132/T) |
| TPDR + 2-CTP → IM46 via TS75 | k(T) = (1.37 × 10−14) exp (−3265/T) |
| TPDR + 2-CTP → IM47 + Cl via TS76 | k(T) = (1.88 × 10−15) exp (−7484/T) |
| TPDR + 2-CTP → IM48 via TS77 | k(T) = (4.21 × 10−14) exp (−6501/T) |
| TPDR + 2-CTP → IM49 + Cl TS78 | k(T) = (7.95 × 10−15) exp (−8890/T) |
| TPDR + 2-CP → IM50 via TS79 | k(T) = (8.93 × 10−14) exp (−3646/T) |
| TPDR + 2-CP → IM51 + Cl via TS80 | k(T) = (9.45 × 10−15) exp (−5768/T) |
| TPDR + 2-CP → IM52 via TS81 | k(T) = (2.02 × 10−14) exp (−6885/T) |
| TPDR + 2-CP → IM53 + Cl TS82 | k(T) = (3.08 × 10−14) exp (−9091/T) |
| C(T)PR1 with 2-C(T)P | C(T)PR2 and (T)PR2 with 2-C(T)P | C(T)PDR and (T)PDR with 2-C(T)P | ||||
|---|---|---|---|---|---|---|
| C/C type | O/C type | S/C type | C/C type | C/C type | O/C type | S/C type |
| 7.28 × 10−23 | 1.75 × 10−20 | 2.60 × 10−16 | 2.34 × 10−16 | 9.82 × 10−16 | 3.24 × 10−20 | 1.59 × 10−17 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zuo, C.; Wang, H.; Pan, W.; Zheng, S.; Xu, F.; Zhang, Q. Quantum Chemical and Kinetic Study on Radical/Molecule Formation Mechanism of Pre-Intermediates for PCTA/PT/DT/DFs from 2-Chlorothiophenol and 2-Chlorophenol Precursors. Int. J. Mol. Sci. 2019, 20, 1542. https://doi.org/10.3390/ijms20071542
Zuo C, Wang H, Pan W, Zheng S, Xu F, Zhang Q. Quantum Chemical and Kinetic Study on Radical/Molecule Formation Mechanism of Pre-Intermediates for PCTA/PT/DT/DFs from 2-Chlorothiophenol and 2-Chlorophenol Precursors. International Journal of Molecular Sciences. 2019; 20(7):1542. https://doi.org/10.3390/ijms20071542
Chicago/Turabian StyleZuo, Chenpeng, Hetong Wang, Wenxiao Pan, Siyuan Zheng, Fei Xu, and Qingzhu Zhang. 2019. "Quantum Chemical and Kinetic Study on Radical/Molecule Formation Mechanism of Pre-Intermediates for PCTA/PT/DT/DFs from 2-Chlorothiophenol and 2-Chlorophenol Precursors" International Journal of Molecular Sciences 20, no. 7: 1542. https://doi.org/10.3390/ijms20071542
APA StyleZuo, C., Wang, H., Pan, W., Zheng, S., Xu, F., & Zhang, Q. (2019). Quantum Chemical and Kinetic Study on Radical/Molecule Formation Mechanism of Pre-Intermediates for PCTA/PT/DT/DFs from 2-Chlorothiophenol and 2-Chlorophenol Precursors. International Journal of Molecular Sciences, 20(7), 1542. https://doi.org/10.3390/ijms20071542