Apoptosis Induction byHistone Deacetylase Inhibitors in Cancer Cells: Role of Ku70


Department of Pharmacology, Shenyang Pharmaceutical University, Shenyang 110016, China
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(7), 1601; https://doi.org/10.3390/ijms20071601
Submission received: 17 January 2019
/
Revised: 16 March 2019
/
Accepted: 18 March 2019
/
Published: 30 March 2019
(This article belongs to the Special Issue Histone Deacetylase Inhibitors in Health and Disease)
Abstract
:Histone deacetylases (HDACs) are a group of enzymes that regulate gene transcription by controlling deacetylation of histones and non-histone proteins. Overexpression of HDACs is found in some types of tumors and predicts poor prognosis. Five HDAC inhibitors are approved for the treatment of cutaneous T-cell lymphoma, peripheral T-cell lymphoma, and multiple myeloma. Treatment with HDAC inhibitors regulates gene expression with increased acetylated histones with unconfirmed connection with therapy. Apoptosis is a key mechanism by which HDAC inhibitors selectively kill cancer cells, probably due to acetylation of non-histone proteins. Ku70 is a protein that repairs DNA breaks and stabilizes anti-apoptotic protein c-FLIP and proapoptotic protein Bax, which is regulated by acetylation. HDAC inhibitors induce Ku70 acetylation with repressed c-FLIP and activated Bax in cancer cells. Current studies indicate that Ku70 is a potential target of HDAC inhibitors and plays an important role during the induction of apoptosis.
1. Introduction
Aberrant acetylation of histone and/or expression of histone deacetylases (HDACs) exist in various solid and hematologic malignancies and contribute to tumorigenesis [1]. Overexpression of individual HDACs accelerates the proliferation of tumor cells and predicts poor prognosis in many types of cancer patients [2,3,4,5,6,7,8]. Genetic knockdown of an individual HDAC, most notably HDAC1, 2, 3, and 6, in different types of tumor cells, such as Hodgkin’s lymphoma, gastric cancer, prostate cancer, colorectal cancer, pancreatic cancer, and ovarian cancer, induced DNA damage, cell cycle arrest, and/or apoptosis [9]. HDACs are being considered as therapeutic targets for cancer treatment. An array of natural and synthetic compounds inhibiting HDAC activity, referred to as HDAC inhibitor (HDACi), have been developed [10,11,12]. Although increased histone acetylation by HDACi treatment has been observed, this phenomenon could not be linked to therapeutic outcome. A series of non-histone proteins including p53, heat shock protein 90 (Hsp90), Akt, tubulin, nuclear factor-k-gene binding (NF-κB), signal transducers and activators of transcription 3 (STAT3), and many others, are also substrates of HDACs [13,14,15]. Cell cycle arrest, cell death, differentiation, autophagy, and senescence account for the antitumor effects of HDACis [10,12]. HDACis induce apoptosis in many types of cancer cells and show relative selectivity to malignant cells. Apoptosis appears to be a key mechanism by which HDACis exert therapeutic effects [16,17,18,19]. Recent studies have shown that HDACi-induced apoptosis involves acetylation of ku70 [20]. The development of HDACis and the potential roles of Ku70 in the HDACi-induced apoptosis are summarized.
2. Classification of HDACs and Cellular Location
Eighteen HDAC subtypes have been found in mammalian cells and are divided into four classes. Class I, II, and IV comprise the “classical” Zn+-dependent HDACs while class III is an NAD+-dependent sirtuin [21,22]. Class I HDACs contain HDAC1, 2, 3, and 8. HDAC1, 2, and 8 are localized in the nucleus while HDAC3 is localized in both the nucleus and cytoplasm. Class II HDACs includes class IIa (HDAC4, 5, 7, 9) and class IIb (HDAC6, 10). Class IIa HDACs shuttle between the nucleus and the cytoplasm when stimulated by different signals while class IIb HDACs is mainly localized in the cytoplasm. Class IV HDACs contain only HDAC11, which has similar features as class I and class II HDACs and is localized in both the nucleus and cytoplasm. Class I HDACs distribute throughout the body, while class II and IV HDACs exhibit tissue-specific expression and distribution in smooth muscle, heart, brain, liver, and colon [23]. Class III HDACs contain seven sirtuin proteins (SIRT1–7) that are expressed in the nucleus, cytoplasm, and mitochondria. Class III HDACs do not share homology with other classes HDACs and are not inhibited by generic HDAC inhibitors [24,25].
3. Biological Functions of HDACs
Deacetylation of histones has been considered as the main function of HDACs. In eukaryotic cells, chromatin is a dense and higher-order structure consisting of DNA and core histone proteins. The nucleosome formed by histone octamers encircling the DNA superhelix is the elementary unit of chromatin [26]. HDACs do not directly bind to DNA but function in transcriptional corepressor complexes with Sin3, nucleosome remodeling deacetylase (NuRD), REST corepressor (CoRest), nuclear receptor co-repressor (N-CoR), and silencing-mediator of retinoic and thyroid receptors (SMRT) [27]. HDACs catalyze deacetylation of the N-terminal lysine residues in core histones and confer a positive charge that enhances the affinity of histones for the negatively charged DNA. Nucleosome histone protein H2A, H2B, H3, and H4 are substrates of HDACs. Hypoacetylation of histones causes chromatin aggregation, which prevents the transcriptional machinery from being close to the DNA and represses gene transcription [28].
HDACs also regulate deacetylation of multiple non-histone protein substrates and impact their functions by altering their activity, cellular location, and protein–protein interactions. More than 50 non-histone proteins have been reported to be the substrates of HDACs. Representatives of non-histone proteins include several important factors, such as p53, NF-κB, STAT3, Hsp90, Akt, and Ku70 (Table 1) [29], which regulate cellular development, proliferation, differentiation, and death and are involved in tumorigenesis (Figure 1).
p53 is a tumor suppressor, and its activity is regulated by acetylation [30]. p53 acetylation blocks ubiquitination and enhances protein stability. Acetylated p53 is an active form and regulates the expression of multiple genes related to cell cycle and apoptosis [30,31,32]. HDAC complexes containing HDAC1 or SIRT1 deacetylate p53 and reverse p53-mediated gene transcription [33,34,35]. HDAC2, 3, 6, and SIRT2 also deacetylate p53 and repress its function [35,36]. p21cip1/waf1 is a transcription target of p53, which can be induced in p53-dependent and independent ways in HDACi-treated cells. p53 has been found to play an important role in HDACi-induced autophagy and apoptosis [37].
NF-κB is a heterodimer of p65 (RelA) and p50 proteins, an inactive complex present in the cytoplasm due to binding to inhibitory proteins known as IκBs [38]. There are multiple acetylation sites on p65 and that acetylation of p65 changes the cellular location of NF-κB [39]. HDAC3 deacetylates p65 and subsequently results in nuclear export to inactivate NF-κB through binding to IκBα [40]. However, different reports show that HDAC3 also positively modulates NF-κB activity by keeping NF-κB in a deacetylated state [41,42,43]. The difference may rely on distinct acetylation sites, which need to be further identified.
Hsp90 serves as a chaperone protein for proper folding and maintenance of a number of proteins including BCR-ABL, mutant Fms-like tyrosine kinase 3 (FLT3), c-Raf, and Akt [44,45,46,47,48]. Hsp90 is usually maintained in a deacetylated form by HDAC6. HDAC6 knockdown or inhibition induces Hsp90 acetylation and results in degradation of the client proteins [49]. HDACi LBH-589 induces Hsp90 acetylation by inhibiting HDAC6 and leads BCR-ABL to dissociation and degradation in leukemic K562 cells [50]. It has been found that knockdown of HDAC3 results in acetylation of Hsp90 in the nucleus and dissociation of DNA damage response (DDR) and homologous recombination (HR) proteins from Hsp90 [51].
The signal transducers and activators of transcription (STATs) belong to a family of cytoplasmic transcription factors and are regulated by phosphorylation and acetylation. Phosphorylated STAT3 translocates to the nucleus to induce gene transcription and to promote cell–cycle progression [52,53]. Acetylated STAT3 locates in the cytoplasm and competitively inhibits nuclear translocation of the phosphorylated form. HDAC3 can bind to and deacetylate STAT3. HDAC3 inhibition increases cytoplasmic acetylation of STAT3 and decreases p-STAT3 protein in the nucleus [54].
Akt is a key factor of the phosphoinositide 3-kinase (PI3K) pathway. Akt could be acetylated by the histone acetyltransferases (HATs) p300 and P300/CBP-associated factor (PCAF) in tumor cell lines [55]; deacetylation of Akt by HDACs is required to maintain its activity. Inhibition of SIRT1 or SIRT2 causes Akt acetylation and, in turn, blocks Akt phosphorylation (active state) [56]. Inhibition of HDAC3 or HDAC6 facilitates Akt acetylation and reduces the level of phosphorylated Akt [57,58].
4. HDACi for Cancer Therapy
A group of small-molecular HDACis has been developed as cancer therapeutics [82]. Four compounds have been approved by the USA FDA and one compound by the China FDA for the treatment of lymphoma and myeloma (Table 2). Vorinostat (SAHA) was the first HDACi approved by the USA FDA for the treatment of cutaneous T-cell lymphoma (CTCL) in 2006 [83]. Romidepsin (FK-228), a natural extraction product derived from Chromobacteriumviolaceum, was subsequently approved in 2009 for the treatment of CTCL and peripheral T-cell lymphoma (PTCL) [84]. Belinostat (PXD-101) was later approved in 2014 for the treatment of relapsed and refractory PTCL [85], and panobinostat (LBH-589) was approved in 2015 for the treatment of relapsed and refractory multiple myeloma [86]. Chidamide (CS055/HBI-8000) was approved by the China FDA in 2015 for the treatment of relapsed or refractory PTCL [87]. SAHA, Belinostat, and LBH-589 are pan-HDACis. Romidepsin is a selective inhibitor of class I HDACs. Chidamide selectively inhibits class I HDACs and HDAC10. None of the five inhibitors inhibit the family of SIRTs and none have been extended to treat other types of cancer. These inhibitors have obvious side effects and toxicities that may restrict their use [88,89,90]. Belinostat and Panobinostat show positive reactions in the Ames mutagenic test, while FK-228 and chidamide have no significant mutagenic toxicity [91,92]. It seems that selective inhibition of class I HDACs is less toxic than inhibition of all HDACs. Specific HDAC inhibitors are being developed [10,93,94,95,96]. Class I/II HDACi Ricolinostat (ACY-1215) and Domatinostat (4SC-202); class I HDACi Mocetinostat (MGCD0103), Entinostat (MS-275), and Banatinostat (CHR3996); and class IIb HDACi Abexinostat (PCI-24781) are being tested in the clinic. To improve the therapeutic effects of HDACis, combinations with radiotherapy, topoisomerase inhibitors, proteasome inhibitors, and BET inhibitors are being tested [11,96].
5. Apoptosis Induction by HDACis
Several mechanisms including cell cycle arrest, differentiation, autophagy, and apoptosis have been reported to mediate the therapeutic effects of HDACis [8,10]. Apoptosis is found to be the most effective manner of killing tumor cells by HDACi [8,97,98,99].
Apoptosis is mediated by extrinsic and intrinsic pathways. The extrinsic pathway is initiated by the binding of death receptors (DR) to specific ligands such as Fas ligand (FasL) and TNF-related apoptosis-inducing ligand (TRAIL). FasL and TRAIL binding leads to trimerization, followed by the recruitment of multiple factors to form a receptor polymer. The adaptor protein Fas-associated death domain protein (FADD) and caspase-8/-10 are recruited to the intracellular tails of the polymer, forming the death-inducing signaling complex (DISC). Caspase-8 and -10 within the DISC facilitate self-activation and activate caspase-3, 6, and/or 7 to trigger apoptosis [100]. FADD-like interleukin 1β-converting enzyme (FLICE) inhibitory protein c-FLIP, which competitively binds to FADD and inhibits caspase-8 activity, is a key negative regulator protein of the extrinsic apoptosis pathway [101]. The intrinsic pathway is controlled mainly by the Bcl-2 family including pro- and anti-apoptotic Bcl-2 family proteins. Anti-apoptotic Bcl-2 proteins include A1, Bcl-2, Bcl-xL, Bcl-w, and Mcl-1. Pro-apoptotic BH3-only protein Bad, Bid, Bim, Bmf, Puma, and Noxa can inhibit anti-apoptotic proteins or activate the effector proteins Bak and Bax [102]. Activated Bak and Bax oligomerize and form membrane pores in the outer membrane of mitochondria, releasing cytochrome c and Smac/Diablo into the cytoplasm, leading to the activation of caspase-9 and caspase-3 and inducing apoptosis [103].
HDACis modulate the balance between pro- and anti-apoptotic proteins and induce both extrinsic and intrinsic apoptotic pathways in cancer cells. Overexpression of prosurvival Bcl-2 family members attenuates HDACi-mediated tumor cell death. HDACis upregulate the BH3-only protein Bim, Bid, and Bmf. HDACis also upregulate DR and TRAIL expression and reduce c-FLIP [104]. Acetylation of non-histone proteins by HDACis may play more important roles in the induction of apoptosis. One such protein is p53, which is acetylated in cancer cells after treatment with HDACi. Acetylated p53 is recruited to the promoters of target genes Bax and Puma to induce their transcription [33,105]. Acetylated p53 also releases the binding to Bax and allows Bax to translocate to the mitochondria to induce apoptosis [106]. Since p53 mutation frequently occurs in many types of cancers that are responsive to HDACi-induced apoptosis, numerous studies suggest that a p53-independent mechanism may be involved in HDACi-mediated apoptosis [107,108,109]. Recent studies indicate that Ku70 is a new protein that regulates apoptosis via acetylation. Other research groups and we have found that HDACi Trichostatin A, SAHA, Nicotinamide, and Tubacin increase acetylation of Ku70 during the apoptosis induction process [16,62,63,110,111].
6. Ku70 Regulates Apoptosis-Related Proteins
Ku70 is a DNA repair factor in the nucleus and forms a heterodimer with Ku80 through the central domain to bind double-strand DNA (dsDNA), which is essential for the classical non-homologous end-joining (c-NHEJ) pathway of double-strand break (DSB) repair [112,113,114].
Many acetylation sites of lysine residues in Ku70, including k539, k542, k544, k553, k556, k317, k331, and k338, have been found. CBP and PCAF have been verified to associate with and acetylate Ku70, which abolishes Ku70 activity for DNA repair and protein interaction [16]. Ku70 is localized not only in the nucleus but is also partially distributed in the cytoplasm [20,115]. Ku70 binds to the cytosolic Bax, Mcl-1, and c-FLIP to increase their stability and protect cells from apoptosis.
6.1. Bax
Bax is one of the two effector proapoptotic proteins to control mitochondrial apoptosis. In cancer cells, Bax exists primarily as a monomer in the cytosol. When given enough stimulation, Bax undergoes conformational changes and translocates to the mitochondria where it oligomerizes and induces apoptosis [116]. Ku70 and Bax were found to interact in the cytoplasm and to inhibit Bax activation [20,64,115,117]. Upon Ku70 knockdown or acetylation, Bax dissociates from the Ku70 complex and translocates to the mitochondria [16,117,118,119]. Overexpression of Ku70 blocks apoptotic cell death induced by transfected Bax [120]. Ku70 possesses enzymatic activity to deubiquitinate its partners and to maintain their stability [121]. Bax association with Ku70 promotes Bax deubiquitylation and prevents its degradation by the proteasome [111,122]. The association with Ku70 also keeps Bax away from the mitochondria [121]. However, Ku70 knockdown showed selective apoptotic effects in neuroblastoma cells but not in HEK293 and Hela cells [16,117]. The difference may rely on the amount of Ku70 bound to Bax and the levels of other antiapoptotic proteins in different cancer cells [115].
6.2. Mcl-1
Mcl-1 is a key anti-apoptotic protein of the Bcl-2 family and inhibits intrinsic apoptosis by inactivation of the effector protein Bak [123]. Wang et al. [124] discovered that Ku70 interacts with Mcl-1 to deubiquitinate and stabilize it. It has been found that the C-terminal tail (aa 536-609) of Ku70 directly deubiquitinates Mcl-1 protein and removes polyubiquitin chains from ubiquitinated Mcl-1. Ku70 deletion promotes Mcl-1 degradation in vitro and in vivo and elevates the sensitivity of lung cancer cells to the Bcl-2 inhibitor ABT-737 [124].
6.3. c-FLIP
Longley et al. found that Ku70 interacts with c-FLIP [125]. Ku70, via its C-terminal region, also binds to and stabilizes c-FLIP protein to prevent its degradation. Acetylation or silencing of Ku70 inhibits its interaction with c-FLIP, leading to c-FLIP degradation by the ubiquitin proteasome system. Downregulation of c-FLIP through acetylation induction of Ku70 could induce apoptosis and/or enhance the sensitivity of cancer cells to chemotherapy [125,126,127].
7. Ku70 Acetylation by HDACi Treatment
HDACi enhances Ku70 acetylation, abolishes DNA repair, increases the levels of DNA damage protein H2AX phosphorylation (γ-H2AX) [128,129,130,131,132], and leads to cell cycle arrest and apoptosis [111,133,134]. MS-275 dramatically elevates Ku70 acetylation and Bax activation in medulloblastoma cells [106]. Maspin inhibits HDAC1, increases acetylation of Ku70, reduces Ku70-mediated sequestration of Bax, and results in Bax-mediated apoptosis [63]. Kwok et al. reported that HDAC6 forms a complex with Ku70 and Bax and that depleting HDAC6 or inhibiting HDAC6 by Tubacin increases Ku70 acetylation and Bax-mediated apoptosis [64]. MS-275 and Tubacin have also been reported to induce c-FLIP degradation [125,127]. HDAC6 is a cytoplasmic HDAC [135,136]. MS-275 is a class I HDACi without inhibiting HDAC6. In class I HDACs, HDAC3, not HDAC1 and 2, locates in both the nucleus and the cytoplasm. It seems that inhibition of HDAC3 should account for the cytoplasmic Ku70 acetylation in MS-275-treated cells. All five approved HDACis inhibit HDAC3 and only three inhibit HDAC6. It is possible that the therapeutic effects of the approved five HDACis come from HDAC3 inhibition but needs to be further confirmed. Currently, several HDAC3 inhibitors are being developed, but there is no clinically useful inhibitor available as yet [93,137].
The currently available HDACis act via inhibiting HDAC activity [138]. Recently, we found that a novel glycyrrhetinic acid derivative 10e reduced the protein levels of HDAC3 and HDAC6, increased Ku70 acetylation, released Bax, and degraded c-FLIP [62]. This compound did not inhibit the activity of HDAC in biochemical assays but increased the levels of acetylated Histone 3. In another report, it was shown that proteasome inhibitor Bortezomib reduced the levels of HDAC1-3 expression and induced histone hyperacetylation and apoptosis [139]. Therefore, selectively degrading an HDAC protein could be an alternative approach for cancer therapy.
8. Summary
Although a series of HDAC inhibitors have been on the market and in clinical development, the death mechanism caused by HDACis is not fully understood. Current evidence demonstrates that apoptosis induction is the most effective way for HDACis to kill tumor cells. Acetylation of non-histone protein substrates seems to play more important roles than regulating histone acetylation in the apoptosis induction process. Ku70 is one of the non-histone protein substrates of HDACs. Ku70 regulates DNA repair and protein stability through direct protein–protein interactions. Ku70 functions as a deubiquitinase to bind to ubiquitinated Bax, Mcl-1, and c-FLIP and to stabilize them (Figure 2). HDAC inhibition, especially HDAC3 and HDAC6, causes cytosolic Ku70 acetylation and dissociation of Bax and c-FLIP, resulting in two proapoptotic programs: Bax activation and c-FLIP degradation, which lead to the initiation of both extrinsic and intrinsic apoptotic programs (Figure 2). Overexpression of Ku70 has been found in various human tumors [140]. It has been found that hepatocellular carcinoma (HCC) patients with high Ku70 expression had an obviously poorer prognosis and lower survival rates than those with low Ku70 expression [141]. Since Ku70 blocks apoptosis, which could be reversed by acetylation, it is an attractive therapeutic target for developing HDACi. More studies are needed to determine which HDAC is preferred for binding to Ku70 and for designing selective HDAC inhibitors.
Author Contributions
P.G. wrote the manuscript; Y.W. contributed to the table and figure design; and Y.J. constructed and edited the manuscript.
Funding
This work was partly supported by The Startup Fund of Shenyang Pharmaceutical University, The Samuel Waxman Cancer Research Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bezecny, P. Histone deacetylase inhibitors in glioblastoma: Pre-clinical and clinical experience. Med. Oncol. 2014, 31, 985. [Google Scholar] [CrossRef]
- Krusche, C.A.; Wulfing, P.; Kersting, C.; Vloet, A.; Bocker, W.; Kiesel, L.; Beier, H.M.; Alfer, J. Histone deacetylase-1 and -3 protein expression in human breast cancer: A tissue microarray analysis. Breast Cancer Res. Treat. 2005, 90, 15–23. [Google Scholar] [CrossRef]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Mitsui, M.; Motoyama, S.; Ogawa, J. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 2011, 74, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Rikimaru, T.; Taketomi, A.; Yamashita, Y.; Shirabe, K.; Hamatsu, T.; Shimada, M.; Maehara, Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology 2007, 72, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Ebert, M.P.; Pross, M.; Dietel, M.; Denkert, C.; Rocken, C. Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: A retrospective analysis. Lancet Oncol. 2008, 9, 139–148. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichert, W.; Roske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Niegisch, G.; Knievel, J.; Koch, A.; Hader, C.; Fischer, U.; Albers, P.; Schulz, W.A. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol. Oncol. 2013, 31, 1770–1779. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.K.; Bishayee, A.; Pandey, A.K. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Wawruszak, A.; Kalafut, J.; Okon, E.; Czapinski, J.; Halasa, M.; Przybyszewska, A.; Miziak, P.; Okla, K.; Rivero-Muller, A.; Stepulak, A. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers 2019, 11, 148. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Yang, X.J.; Seto, E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar] [CrossRef]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.Y.; Lavu, S.; Bitterman, K.J.; Hekking, B.; Imahiyerobo, T.A.; Miller, C.; Frye, R.; Ploegh, H.; Kessler, B.M.; Sinclair, D.A. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 2004, 13, 627–638. [Google Scholar] [CrossRef]
- Costanzo, A.; Merlo, P.; Pediconi, N.; Fulco, M.; Sartorelli, V.; Cole, P.A.; Fontemaggi, G.; Fanciulli, M.; Schiltz, L.; Blandino, G.; et al. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol. Cell 2002, 9, 175–186. [Google Scholar] [CrossRef]
- Huang, B.H.; Laban, M.; Leung, C.H.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Olaharski, A.J.; Rine, J.; Marshall, B.L.; Babiarz, J.; Zhang, L.; Verdin, E.; Smith, M.T. The flavoring agent dihydrocoumarin reverses epigenetic silencing and inhibits sirtuin deacetylases. PLoS Genet. 2005, 1, e77. [Google Scholar] [CrossRef] [PubMed]
- Hada, M.; Kwok, R.P. Regulation of ku70-bax complex in cells. J. Cell Death 2014, 7, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Groselj, B.; Sharma, N.L.; Hamdy, F.C.; Kerr, M.; Kiltie, A.E. Histone deacetylase inhibitors as radiosensitisers: Effects on DNA damage signalling and repair. Br. J. Cancer 2013, 108, 748–754. [Google Scholar] [CrossRef]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.C.; Denu, J.M. Chemical mechanisms of histone lysine and arginine modifications. Biochim. Biophys. Acta 2009, 1789, 45–57. [Google Scholar] [CrossRef]
- Glass, C.K.; Rosenfeld, M.G. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000, 14, 121–141. [Google Scholar]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Dimitroulis, D.; Spartalis, E.; Margonis, G.A.; Schizas, D.; Deskou, I.; Doula, C.; Magkouti, E.; et al. Targeting Histone Deacetylases in Malignant Melanoma: A Future Therapeutic Agent or Just Great Expectations? Anticancer Res. 2017, 37, 5355–5362. [Google Scholar] [PubMed]
- Singh, B.N.; Zhang, G.; Hwa, Y.L.; Li, J.; Dowdy, S.C.; Jiang, S.W. Nonhistone protein acetylation as cancer therapy targets. Expert Rev. Anticancer Ther. 2010, 10, 935–954. [Google Scholar] [CrossRef] [Green Version]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Ding, G.; Liu, H.D.; Huang, Q.; Liang, H.X.; Ding, Z.H.; Liao, Z.J.; Huang, G. HDAC6 promotes hepatocellular carcinoma progression by inhibiting P53 transcriptional activity. FEBS Lett. 2013, 587, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Mrakovcic, M.; Bohner, L.; Hanisch, M.; Frohlich, L.F. Epigenetic Targeting of Autophagy via HDAC Inhibition in Tumor Cells: Role of p53. Int. J. Mol. Sci. 2018, 19, 3952. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.E.; Ghosh, G. Regulation of DNA binding by Rel/NF-kappaB transcription factors: Structural views. Oncogene 1999, 18, 6845–6852. [Google Scholar] [CrossRef]
- Ito, K. Impact of post-translational modifications of proteins on the inflammatory process. Biochem. Soc. Trans. 2007, 35, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef]
- Ashburner, B.P.; Westerheide, S.D.; Baldwin, A.S., Jr. The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell. Biol. 2001, 21, 7065–7077. [Google Scholar] [CrossRef] [PubMed]
- Imre, G.; Gekeler, V.; Leja, A.; Beckers, T.; Boehm, M. Histone deacetylase inhibitors suppress the inducibility of nuclear factor-kappaB by tumor necrosis factor-alpha receptor-1 down-regulation. Cancer Res. 2006, 66, 5409–5418. [Google Scholar] [CrossRef]
- Takada, Y.; Gillenwater, A.; Ichikawa, H.; Aggarwal, B.B. Suberoylanilide hydroxamic acid potentiates apoptosis, inhibits invasion, and abolishes osteoclastogenesis by suppressing nuclear factor-kappaB activation. J. Biol. Chem. 2006, 281, 5612–5622. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Hayer-Hartl, M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science 2002, 295, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, J.S.; Xu, W.; Neckers, L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 2003, 3, 213–217. [Google Scholar] [CrossRef] [Green Version]
- Nimmanapalli, R.; O’Bryan, E.; Bhalla, K. Geldanamycin and its analogue 17-allylamino-17-demethoxygeldanamycin lowers Bcr-Abl levels and induces apoptosis and differentiation of Bcr-Abl-positive human leukemic blasts. Cancer Res. 2001, 61, 1799–1804. [Google Scholar]
- George, P.; Bali, P.; Cohen, P.; Tao, J.; Guo, F.; Sigua, C.; Vishvanath, A.; Fiskus, W.; Scuto, A.; Annavarapu, S.; et al. Cotreatment with 17-allylamino-demethoxygeldanamycin and FLT-3 kinase inhibitor PKC412 is highly effective against human acute myelogenous leukemia cells with mutant FLT-3. Cancer Res. 2004, 64, 3645–3652. [Google Scholar] [CrossRef] [PubMed]
- Bagatell, R.; Whitesell, L. Altered Hsp90 function in cancer: A unique therapeutic opportunity. Mol. Cancer Ther. 2004, 3, 1021–1030. [Google Scholar]
- Kramer, O.H.; Mahboobi, S.; Sellmer, A. Drugging the HDAC6-HSP90 interplay in malignant cells. Trends Pharmacol. Sci. 2014, 35, 501–509. [Google Scholar] [CrossRef] [PubMed]
- George, P.; Bali, P.; Annavarapu, S.; Scuto, A.; Fiskus, W.; Guo, F.; Sigua, C.; Sondarva, G.; Moscinski, L.; Atadja, P.; et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17-AAG is highly active against human CML-BC cells and AML cells with activating mutation of FLT-3. Blood 2005, 105, 1768–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M.; et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 2014, 5, 5637–5650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Han, J.J.; Stenson, M.; Wellik, L.; Witzig, T.E. Regulation of STAT3 by histone deacetylase-3 in diffuse large B-cell lymphoma: Implications for therapy. Leukemia 2012, 26, 1356–1364. [Google Scholar] [CrossRef]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.; Gupta, M.P. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 2011, 4, ra46. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Sundaresan, N.R.; Gupta, M.P. Regulation of Akt signaling by sirtuins: Its implication in cardiac hypertrophy and aging. Circ. Res. 2014, 114, 368–378. [Google Scholar] [CrossRef]
- Iaconelli, J.; Lalonde, J.; Watmuff, B.; Liu, B.; Mazitschek, R.; Haggarty, S.J.; Karmacharya, R. Lysine Deacetylation by HDAC6 Regulates the Kinase Activity of AKT in Human Neural Progenitor Cells. ACS Chem. Biol. 2017, 12, 2139–2148. [Google Scholar] [CrossRef]
- Long, J.; Fang, W.Y.; Chang, L.; Gao, W.H.; Shen, Y.; Jia, M.Y.; Zhang, Y.X.; Wang, Y.; Dou, H.B.; Zhang, W.J.; et al. Targeting HDAC3, a new partner protein of AKT in the reversal of chemoresistance in acute myeloid leukemia via DNA damage response. Leukemia 2017, 31, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef]
- Rothgiesser, K.M.; Erener, S.; Waibel, S.; Luscher, B.; Hottiger, M.O. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010, 123, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ye, J.; Chen, X.; Shi, J.; Wu, W.; Lin, W.; Lin, W.; Li, Y.; Fu, H.; Li, S. Valproic acid attenuates traumatic spinal cord injury-induced inflammation via STAT1 and NF-kappaB pathway dependent of HDAC3. J. Neuroinflamm. 2018, 15, 150. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Li, K.; Li, Y.; Liu, D.; Zhao, L.; Jing, Y. HDAC and Ku70 axis- an effective target for apoptosis induction by a new 2-cyano-3-oxo-1,9-dien glycyrrhetinic acid analogue. Cell Death Dis. 2018, 9, 623. [Google Scholar] [CrossRef]
- Lee, S.J.; Jang, H.; Park, C. Maspin increases Ku70 acetylation and Bax-mediated cell death in cancer cells. Int. J. Mol. Med. 2012, 29, 225–230. [Google Scholar]
- Subramanian, C.; Jarzembowski, J.A.; Opipari, A.W., Jr.; Castle, V.P.; Kwok, R.P. HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia 2011, 13, 726–734. [Google Scholar] [CrossRef]
- Roth, M.; Wang, Z.; Chen, W.Y. SIRT1 and LSD1 competitively regulate KU70 functions in DNA repair and mutation acquisition in cancer cells. Oncotarget 2016, 7, 50195–50214. [Google Scholar] [CrossRef] [Green Version]
- Sundaresan, N.R.; Samant, S.A.; Pillai, V.B.; Rajamohan, S.B.; Gupta, M.P. SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 2008, 28, 6384–6401. [Google Scholar] [CrossRef]
- Tao, N.N.; Ren, J.H.; Tang, H.; Ran, L.K.; Zhou, H.Z.; Liu, B.; Huang, A.L.; Chen, J. Deacetylation of Ku70 by SIRT6 attenuates Bax-mediated apoptosis in hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2017, 485, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820–6831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Zhuang, Q.; Li, W.; Benda, C.; Huang, Z.; Ahmed, T.; Liu, P.; Guo, X.; Ibanez, D.P.; Luo, Z.; Zhang, M.; et al. NCoR/SMRT co-repressors cooperate with c-MYC to create an epigenetic barrier to somatic cell reprogramming. Nat. Cell Biol. 2018, 20, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, X.; Polakiewicz, R.D.; Yao, T.P.; Comb, M.J. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Biol. Chem. 2008, 283, 12686–12690. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Sue, Y.M.; Yang, N.J.; Lee, Y.H.; Juan, S.H. 3-Methylcholanthrene, an AhR agonist, caused cell-cycle arrest by histone deacetylation through a RhoA-dependent recruitment of HDAC1 and pRb2 to E2F1 complex. PLoS ONE 2014, 9, e92793. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Yang, F.Q.; Liu, M.; Yang, F.P.; Che, J.; Li, W.; Zhai, W.; Wang, G.C.; Zheng, J.H.; Li, X. VPA inhibits renal cancer cell migration by targeting HDAC2 and down-regulating HIF-1alpha. Mol. Biol. Rep. 2014, 41, 1511–1518. [Google Scholar] [CrossRef]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J. Biol. Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef]
- Ye, M.; Fang, Z.; Gu, H.; Song, R.; Ye, J.; Li, H.; Wu, Z.; Zhou, S.; Li, P.; Cai, X.; et al. Histone deacetylase 5 promotes the migration and invasion of hepatocellular carcinoma via increasing the transcription of hypoxia-inducible factor-1alpha under hypoxia condition. Tumour Biol. 2017, 39, 1010428317705034. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Li, H.; Avraham, S.; Jiang, S.; Avraham, H.K. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor alpha. Int. J. Cancer 2003, 107, 353–358. [Google Scholar] [CrossRef]
- Iaconelli, J.; Huang, J.H.; Berkovitch, S.S.; Chattopadhyay, S.; Mazitschek, R.; Schreiber, S.L.; Haggarty, S.J.; Karmacharya, R. HDAC6 inhibitors modulate Lys49 acetylation and membrane localization of beta-catenin in human iPSC-derived neuronal cells. ACS Chem. Biol. 2015, 10, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, Y.; Towatari, M.; Tsuzuki, S.; Hayakawa, F.; Maeda, T.; Miyata, Y.; Tanimoto, M.; Saito, H. Histone deacetylase 3 associates with and represses the transcription factor GATA-2. Blood 2001, 98, 2116–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watamoto, K.; Towatari, M.; Ozawa, Y.; Miyata, Y.; Okamoto, M.; Abe, A.; Naoe, T.; Saito, H. Altered interaction of HDAC5 with GATA-1 during MEL cell differentiation. Oncogene 2003, 22, 9176–9184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Horst, A.; Tertoolen, L.G.; de Vries-Smits, L.M.; Frye, R.A.; Medema, R.H.; Burgering, B.M. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J. Biol. Chem. 2004, 279, 28873–28879. [Google Scholar] [CrossRef]
- Zhang, H.; Shang, Y.P.; Chen, H.Y.; Li, J. Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatol. Res. 2017, 47, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncology 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef]
- Sawas, A.; Radeski, D.; O’Connor, O.A. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: A perspective review. Ther. Adv. Hematol. 2015, 6, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Moore, D. Panobinostat (Farydak): A Novel Option for the Treatment of Relapsed Or Relapsed and Refractory Multiple Myeloma. Pharm. Ther. 2016, 41, 296–300. [Google Scholar]
- Lu, X.; Ning, Z.; Li, Z.; Cao, H.; Wang, X. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis. Res. 2016, 5, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef]
- Mackay, H.J.; Hirte, H.; Colgan, T.; Covens, A.; MacAlpine, K.; Grenci, P.; Wang, L.; Mason, J.; Pham, P.A.; Tsao, M.S.; et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur. J. Cancer 2010, 46, 1573–1579. [Google Scholar] [CrossRef]
- Morita, S.; Oizumi, S.; Minami, H.; Kitagawa, K.; Komatsu, Y.; Fujiwara, Y.; Inada, M.; Yuki, S.; Kiyota, N.; Mitsuma, A.; et al. Phase I dose-escalating study of panobinostat (LBH589) administered intravenously to Japanese patients with advanced solid tumors. Investig. New Drugs 2012, 30, 1950–1957. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.S.; Galloway, S.; Lagrutta, A.; Armstrong, M.; Miller, T.; Richon, V.M.; Andrews, P.A. Nonclinical safety assessment of the histone deacetylase inhibitor vorinostat. Int. J. Toxicol. 2010, 29, 3–19. [Google Scholar] [CrossRef]
- Shen, S.; Kozikowski, A.P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors--What Some May Have Forgotten or Would Rather Forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Zwinderman, M.R.H.; Dekker, F.J. The Process and Strategy for Developing Selective Histone Deacetylase 3 Inhibitors. Molecules 2018, 23, 551. [Google Scholar]
- Dallavalle, S.; Pisano, C.; Zunino, F. Development and therapeutic impact of HDAC6-selective inhibitors. Biochem. Pharm. 2012, 84, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Kaletsch, A.; Pinkerneil, M.; Hoffmann, M.J.; Jaguva Vasudevan, A.A.; Wang, C.; Hansen, F.K.; Wiek, C.; Hanenberg, H.; Gertzen, C.; Gohlke, H.; et al. Effects of novel HDAC inhibitors on urothelial carcinoma cells. Clin. Epigenet. 2018, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Manzotti, G.; Ciarrocchi, A.; Sancisi, V. Inhibition of BET Proteins and Histone Deacetylase (HDACs): Crossing Roads in Cancer Therapy. Cancers 2019, 11, 304. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.P.; Singh, M.M.; Rivera-Del Valle, N.; Manton, C.A.; Chandra, J. Therapeutic strategies to enhance the anticancer efficacy of histone deacetylase inhibitors. J. Biomed. Biotechnol. 2011, 2011, 514261. [Google Scholar] [CrossRef]
- Roy, S.; Nicholson, D.W. Cross-talk in cell death signaling. J. Exp. Med. 2000, 192, 21–26. [Google Scholar] [CrossRef]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2009, 124, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, D.; Tu, H.C.; Kim, H.; Wang, G.X.; Bean, G.R.; Takeuchi, O.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.; Cheng, E.H. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 2010, 330, 1390–1393. [Google Scholar] [CrossRef]
- Elkholi, R.; Floros, K.V.; Chipuk, J.E. The Role of BH3-Only Proteins in Tumor Cell Development, Signaling, and Treatment. Genes Cancer 2011, 2, 523–537. [Google Scholar] [CrossRef] [Green Version]
- Matthews, G.M.; Newbold, A.; Johnstone, R.W. Intrinsic and extrinsic apoptotic pathway signaling as determinants of histone deacetylase inhibitor antitumor activity. Adv. Cancer Res. 2012, 116, 165–197. [Google Scholar] [PubMed]
- Sasca, D.; Hahnel, P.S.; Szybinski, J.; Khawaja, K.; Kriege, O.; Pante, S.V.; Bullinger, L.; Strand, S.; Strand, D.; Theobald, M.; et al. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood 2014, 124, 121–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacker, S.; Karl, S.; Mader, I.; Cristofanon, S.; Schweitzer, T.; Krauss, J.; Rutkowski, S.; Debatin, K.M.; Fulda, S. Histone deacetylase inhibitors prime medulloblastoma cells for chemotherapy-induced apoptosis by enhancing p53-dependent Bax activation. Oncogene 2011, 30, 2275–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, C.; Ikezoe, T.; Yang, J.; Udaka, K.; Yokoyama, A. Simultaneous inhibition of DNA methyltransferase and histone deacetylase induces p53-independent apoptosis via down-regulation of Mcl-1 in acute myelogenous leukemia cells. Leuk. Res. 2011, 35, 932–939. [Google Scholar] [CrossRef]
- Hirose, T.; Sowa, Y.; Takahashi, S.; Saito, S.; Yasuda, C.; Shindo, N.; Furuichi, K.; Sakai, T. p53-independent induction of Gadd45 by histone deacetylase inhibitor: Coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene 2003, 22, 7762–7773. [Google Scholar] [CrossRef] [PubMed]
- Vrana, J.A.; Decker, R.H.; Johnson, C.R.; Wang, Z.; Jarvis, W.D.; Richon, V.M.; Ehinger, M.; Fisher, P.B.; Grant, S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene 1999, 18, 7016–7025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucherat, O.; Chabot, S.; Paulin, R.; Trinh, I.; Bourgeois, A.; Potus, F.; Lampron, M.C.; Lambert, C.; Breuils-Bonnet, S.; Nadeau, V.; et al. HDAC6: A Novel Histone Deacetylase Implicated in Pulmonary Arterial Hypertension. Sci. Rep. 2017, 7, 4546. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Zhang, F.; Zhang, X.T.; Zhang, T.; Li, Y.H.; Fan, L.; Sun, Y.; Zhang, H.L.; Mei, Q.B. Ku70 is essential for histone deacetylase inhibitor trichostatin A-induced apoptosis. Mol. Med. Rep. 2015, 12, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gao, F.; Wang, T.; Flagg, T.; Deng, X. A nonhomologous end-joining pathway is required for protein phosphatase 2A promotion of DNA double-strand break repair. Neoplasia 2009, 11, 1012–1021. [Google Scholar] [CrossRef]
- Li, Z.; Owonikoko, T.K.; Sun, S.Y.; Ramalingam, S.S.; Doetsch, P.W.; Xiao, Z.Q.; Khuri, F.R.; Curran, W.J.; Deng, X. c-Myc suppression of DNA double-strand break repair. Neoplasia 2012, 14, 1190–1202. [Google Scholar] [CrossRef]
- Hada, M.; Subramanian, C.; Andrews, P.C.; Kwok, R.P. Cytosolic Ku70 regulates Bax-mediated cell death. Tumour Biol. 2016, 37, 13903–13914. [Google Scholar] [CrossRef] [Green Version]
- Antignani, A.; Youle, R.J. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr. Opin. Cell Biol. 2006, 18, 685–689. [Google Scholar] [CrossRef]
- Subramanian, C.; Opipari, A.W., Jr.; Bian, X.; Castle, V.P.; Kwok, R.P. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 4842–4847. [Google Scholar] [CrossRef] [Green Version]
- Ngo, J.; Matsuyama, M.; Kim, C.; Poventud-Fuentes, I.; Bates, A.; Siedlak, S.L.; Lee, H.G.; Doughman, Y.Q.; Watanabe, M.; Liner, A.; et al. Bax deficiency extends the survival of Ku70 knockout mice that develop lung and heart diseases. Cell Death Dis. 2015, 6, e1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuyama, S.; Palmer, J.; Bates, A.; Poventud-Fuentes, I.; Wong, K.; Ngo, J.; Matsuyama, M. Bax-induced apoptosis shortens the life span of DNA repair defect Ku70-knockout mice by inducing emphysema. Exp. Biol. Med. 2016, 241, 1265–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishnudas, V.K.; Miller, J.B. Ku70 regulates Bax-mediated pathogenesis in laminin-alpha2-deficient human muscle cells and mouse models of congenital muscular dystrophy. Hum. Mol. Genet. 2009, 18, 4467–4477. [Google Scholar] [CrossRef]
- Amsel, A.D.; Rathaus, M.; Kronman, N.; Cohen, H.Y. Regulation of the proapoptotic factor Bax by Ku70-dependent deubiquitylation. Proc. Natl. Acad. Sci. USA 2008, 105, 5117–5122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathaus, M.; Lerrer, B.; Cohen, H.Y. DeubiKuitylation: A novel DUB enzymatic activity for the DNA repair protein, Ku70. Cell Cycle 2009, 8, 1843–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perciavalle, R.M.; Opferman, J.T. Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol. 2013, 23, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xie, M.; Li, R.; Owonikoko, T.K.; Ramalingam, S.S.; Khuri, F.R.; Curran, W.J.; Wang, Y.; Deng, X. Role of Ku70 in deubiquitination of Mcl-1 and suppression of apoptosis. Cell Death Differ. 2014, 21, 1160–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, E.; Holohan, C.; McLaughlin, K.M.; Majkut, J.; Dolan, S.; Redmond, K.; Riley, J.; McLaughlin, K.; Stasik, I.; Crudden, M.; et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ. 2012, 19, 1317–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLornan, D.; Hay, J.; McLaughlin, K.; Holohan, C.; Burnett, A.K.; Hills, R.K.; Johnston, P.G.; Mills, K.I.; McMullin, M.F.; Longley, D.B.; et al. Prognostic and therapeutic relevance of c-FLIP in acute myeloid leukaemia. Br. J. Haematol. 2013, 160, 188–198. [Google Scholar] [CrossRef]
- McCann, C.; Crawford, N.; Majkut, J.; Holohan, C.; Armstrong, C.W.D.; Maxwell, P.J.; Ong, C.W.; LaBonte, M.J.; McDade, S.S.; Waugh, D.J.; et al. Cytoplasmic FLIP(S) and nuclear FLIP(L) mediate resistance of castrate-resistant prostate cancer to apoptosis induced by IAP antagonists. Cell Death Dis. 2018, 9, 1081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wu, H.; Yang, M.; Ye, S.; Li, L.; Zhang, H.; Hu, J.; Wang, X.; Xu, J.; Liang, A. SIRT1 inhibition impairs non-homologous end joining DNA damage repair by increasing Ku70 acetylation in chronic myeloid leukemia cells. Oncotarget 2016, 7, 13538–13550. [Google Scholar] [CrossRef] [PubMed]
- Valdez, B.C.; Li, Y.; Murray, D.; Liu, Y.; Nieto, Y.; Champlin, R.E.; Andersson, B.S. Combination of a hypomethylating agent and inhibitors of PARP and HDAC traps PARP1 and DNMT1 to chromatin, acetylates DNA repair proteins, down-regulates NuRD and induces apoptosis in human leukemia and lymphoma cells. Oncotarget 2018, 9, 3908–3921. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Nagaria, P.K.; Pawar, N.; Adewuyi, A.; Gojo, I.; Meyers, D.J.; Cole, P.A.; Rassool, F.V. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk. Res. 2016, 45, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makita, N.; Ninomiya, I.; Tsukada, T.; Okamoto, K.; Harada, S.; Nakanuma, S.; Sakai, S.; Makino, I.; Kinoshita, J.; Hayashi, H.; et al. Inhibitory effects of valproic acid in DNA double-strand break repair after irradiation in esophageal squamous carcinoma cells. Oncol. Rep. 2015, 34, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Rosato, R.R.; Almenara, J.A.; Maggio, S.C.; Coe, S.; Atadja, P.; Dent, P.; Grant, S. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol. Cancer Ther. 2008, 7, 3285–3297. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Wang, H.; Xu, S.; Han, P.; Xin, M.; Zhou, J.L. Sensitization of suberoylanilide hydroxamic acid (SAHA) on chemoradiation for human cervical cancer cells and its mechanism. Eur. J. Gynaecol. Oncol. 2015, 36, 117–122. [Google Scholar] [PubMed]
- Busser, B.; Sancey, L.; Josserand, V.; Niang, C.; Khochbin, S.; Favrot, M.C.; Coll, J.L.; Hurbin, A. Amphiregulin promotes resistance to gefitinib in nonsmall cell lung cancer cells by regulating Ku70 acetylation. Mol. Ther. 2010, 18, 536–543. [Google Scholar] [CrossRef]
- Di Giorgio, E.; Gagliostro, E.; Brancolini, C. Selective class IIa HDAC inhibitors: Myth or reality. Cell. Mol. Life Sci. 2015, 72, 73–86. [Google Scholar] [CrossRef]
- Valenzuela-Fernandez, A.; Cabrero, J.R.; Serrador, J.M.; Sanchez-Madrid, F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008, 18, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, N.; Amin, S.A.; Trivedi, P.; Jha, T.; Ghosh, B. HDAC3 is a potential validated target for cancer: An overview on the benzamide-based selective HDAC3 inhibitors through comparative SAR/QSAR/QAAR approaches. Eur. J. Med. Chem. 2018, 157, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, J.; Wada, T.; Shimizu, R.; Izumi, T.; Akutsu, M.; Mitsunaga, K.; Noborio-Hatano, K.; Nobuyoshi, M.; Ozawa, K.; Kano, Y.; et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood 2010, 116, 406–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshareeda, A.T.; Negm, O.H.; Albarakati, N.; Green, A.R.; Nolan, C.; Sultana, R.; Madhusudan, S.; Benhasouna, A.; Tighe, P.; Ellis, I.O.; et al. Clinicopathological significance of KU70/KU80, a key DNA damage repair protein in breast cancer. Breast Cancer Res. Treat. 2013, 139, 301–310. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, X.; Shi, W.; Xu, J.; Fan, H.; Zhang, S.; Ni, R. The DNA damage repair protein Ku70 regulates tumor cell and hepatic carcinogenesis by interacting with FOXO4. Pathol. Res. Pract. 2016, 212, 153–161. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Non-histone protein substrates of histone deacetylases (HDACs) regulate tumor cell proliferation, cycling, apoptosis, and DNA damage repair. GSK3β, glycogen synthase kinase3β; DSB, double-strand break.
Figure 1.
Non-histone protein substrates of histone deacetylases (HDACs) regulate tumor cell proliferation, cycling, apoptosis, and DNA damage repair. GSK3β, glycogen synthase kinase3β; DSB, double-strand break.

Figure 2.
Cytoplasmic Ku70 regulates the activity and stability of apoptosis-related proteins via acetylation. Cytoplasmic Ku70 as a deubiquitinase binds to ubiquitinated Bax, c-FLIP, and Mcl-1 to prevent them from being degraded by proteasomes. After treatment with HDACi, Ku70 is acetylated and dissociates Bax and c-FLIP from the complexes, leading to Bax activation, c-FLIP degradation, and apoptosis initiation. It has been reported that inhibition of HDAC1, HDAC3, HDAC6, SIRT1, SIRT3, or SIRT6 induces Ku70-mediated Bax activation [62,63,64,65,66,67] and that inhibition of HDAC3 and HDAC6 induces Ku70-mediated c-FLIP degradation [62,125].
Figure 2.
Cytoplasmic Ku70 regulates the activity and stability of apoptosis-related proteins via acetylation. Cytoplasmic Ku70 as a deubiquitinase binds to ubiquitinated Bax, c-FLIP, and Mcl-1 to prevent them from being degraded by proteasomes. After treatment with HDACi, Ku70 is acetylated and dissociates Bax and c-FLIP from the complexes, leading to Bax activation, c-FLIP degradation, and apoptosis initiation. It has been reported that inhibition of HDAC1, HDAC3, HDAC6, SIRT1, SIRT3, or SIRT6 induces Ku70-mediated Bax activation [62,63,64,65,66,67] and that inhibition of HDAC3 and HDAC6 induces Ku70-mediated c-FLIP degradation [62,125].


{kind=link}
{kind=link}
Table 1.
Non-histone proteins deacetylated by histone deacetylases (HDACs).
| Substrate | HDACs | Function of Acetylation | Reference |
|---|---|---|---|
| p53 | HDAC1, 2, 3, 6, SIRT1, 2, 7 | Increases transcription activity | [33,34,35,36] |
| NF-κB | HDAC3, SIRT1, 2 | Impacts NF-κB translocation and NF-κB-mediated inflammation and transcription | [39,40,59,60] |
| Hsp90 | HDAC3, 6 | Promotes chaperone protein degradation | [49,50,51] |
| STATs | HDAC3 | Inhibits JAK2/STAT pathway and NF-kB-mediated inflammatory response | [54,61] |
| Akt | HDAC3, 6, SIRT1, 2 | Decreases Akt phosphorylation and inhibits Akt-mediated pathway | [55,56,57,58] |
| Ku70 | HDAC1, 3, 6, SIRT1, 3, 6 | Enhances DNA damage and reduces the stability of Ku70-regulated proteins | [62,63,64,65,66,67] |
| α-tubulin | HDAC6, SIRT2 | Inhibits microtubule assembly and cell motility | [68,69] |
| c-Myc | HDAC3, 6 | Inhibits the transcription ability and decreases c-Myc expression | [70,71] |
| E2F1 | HDAC1 | Increases DNA-binding affinity | [72] |
| HIF-1α | HDAC2, 4, 5, 7 | Regulates its degradation | [73,74,75,76] |
| ER | HDAC1 | Increases cell proliferation | [77] |
| β-catenin | HDAC6 | Inhibits its nuclear translocation | [78] |
| GATAs | HDAC3, 4, 5 | Affects erythroid differentiation | [79,80] |
| FoxOs | SIRT1 | Decreases DNA-binding affinity and reduces transcription activity | [81] |
Table 2.
The HDAC inhibitors approved for clinic use.
| HDAC Inhibitor | Chemical Class | Structure of the Compound | HDAC Class Inhibited |
|---|---|---|---|
| Vorinostat (SAHA) | Hydroxamic acid | 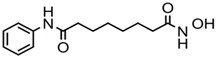 | Pan-HDACs |
| Romidepsin (FK228) | Cyclic peptide |  | Class I |
| Belinostat (PXD-101) | Hydroxamic acid |  | Pan-HDACs |
| Panobinostat (LBH-589) | Hydroxamic acid | 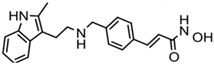 | Pan-HDACs |
| Chidamide | Benzamide |  | Class I, HDAC10 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gong, P.; Wang, Y.; Jing, Y. Apoptosis Induction byHistone Deacetylase Inhibitors in Cancer Cells: Role of Ku70. Int. J. Mol. Sci. 2019, 20, 1601. https://doi.org/10.3390/ijms20071601
AMA Style
Gong P, Wang Y, Jing Y. Apoptosis Induction byHistone Deacetylase Inhibitors in Cancer Cells: Role of Ku70. International Journal of Molecular Sciences. 2019; 20(7):1601. https://doi.org/10.3390/ijms20071601
Chicago/Turabian StyleGong, Ping, Yuetong Wang, and Yongkui Jing. 2019. "Apoptosis Induction byHistone Deacetylase Inhibitors in Cancer Cells: Role of Ku70" International Journal of Molecular Sciences 20, no. 7: 1601. https://doi.org/10.3390/ijms20071601
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.