Mitochondrial Targeting of Antioxidants Alters Pancreatic Acinar Cell Bioenergetics and Determines Cell Fate

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
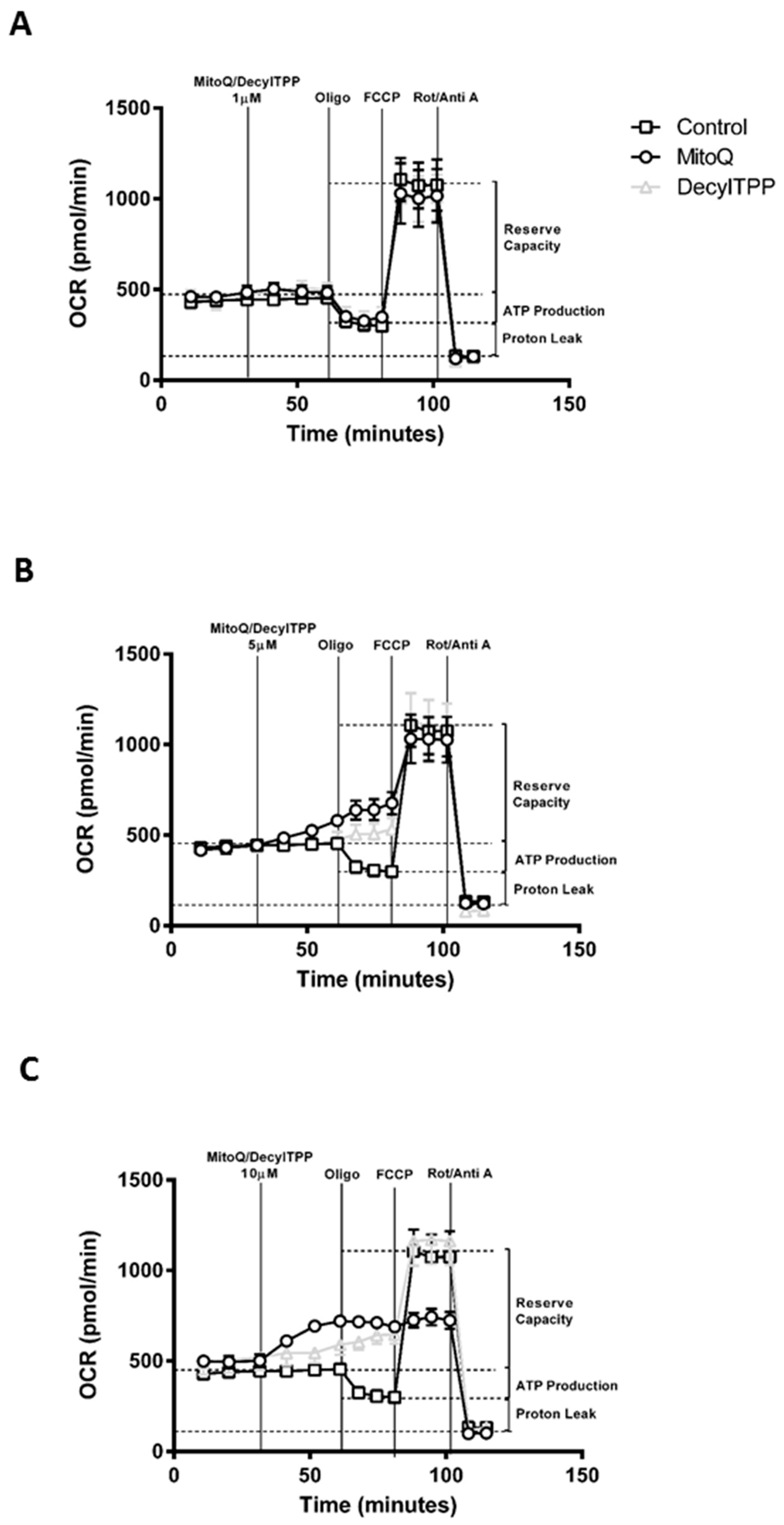
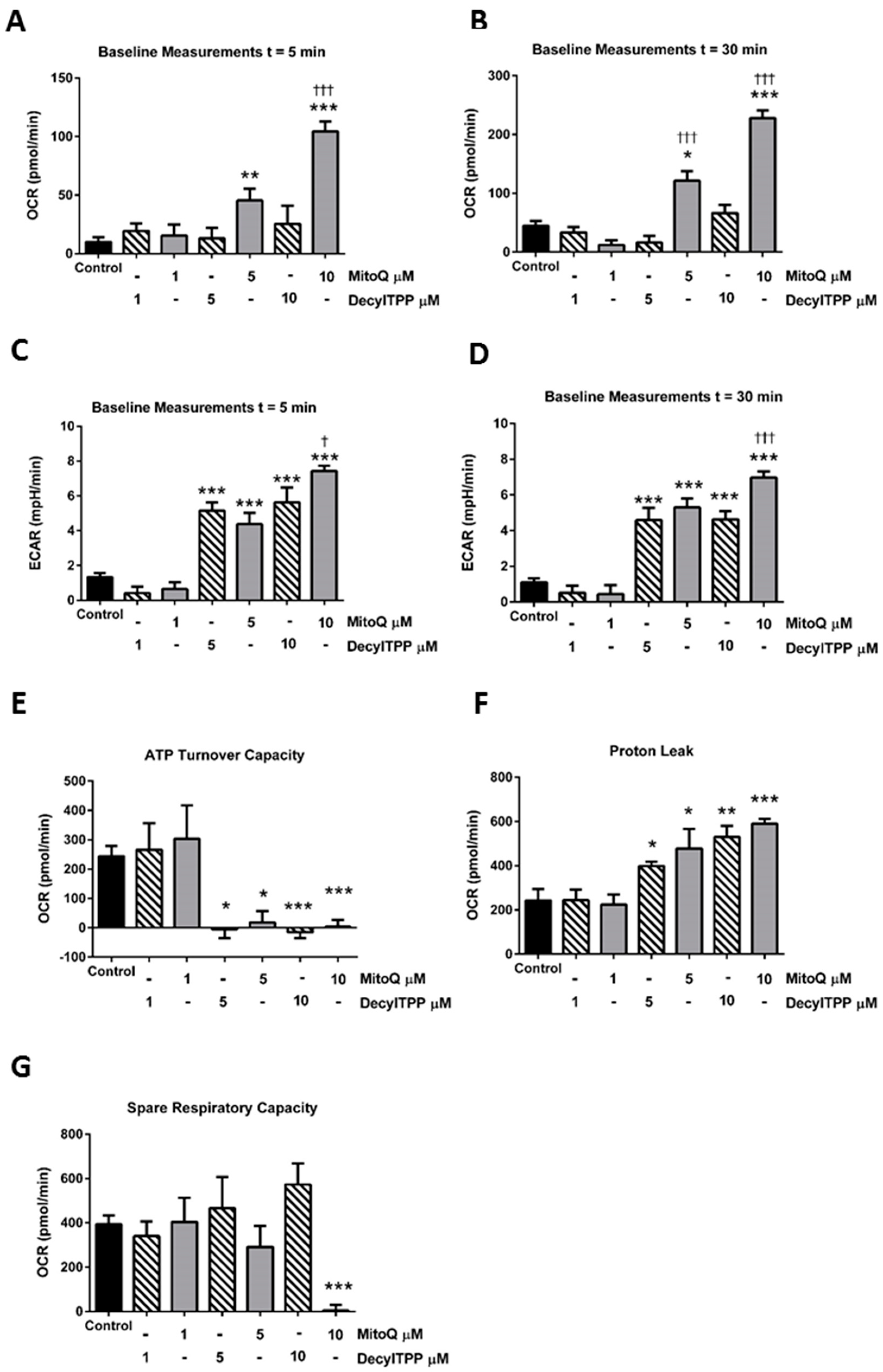
2.1. Differential Effects of MitoQ and DecylTPP on Acinar Cell Bioenergetics
2.2. Inhibitory Effects of MitoQ on the NADH/FAD+ Redox Ratio
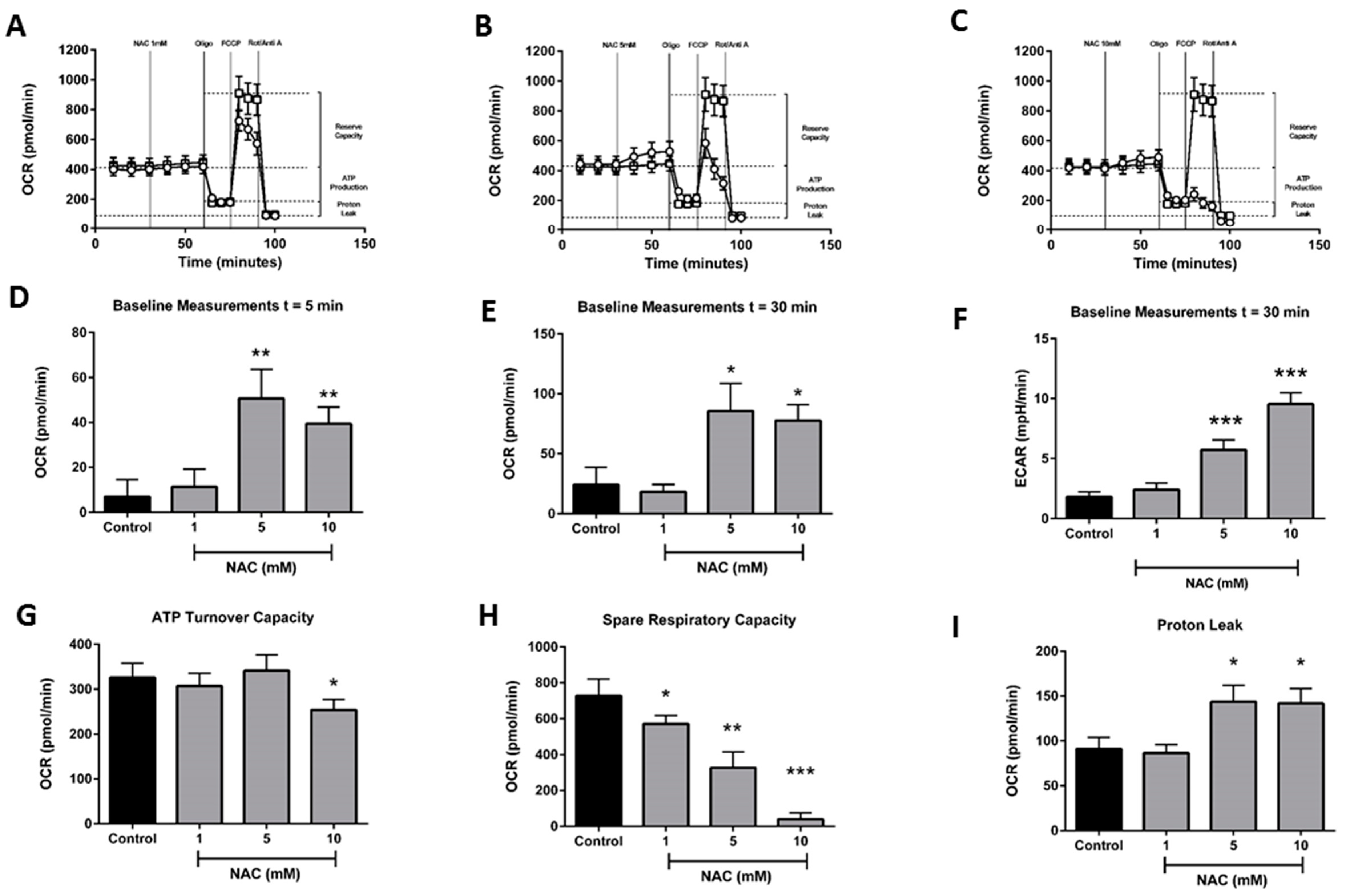
2.3. Effects of N-Acetylcysteine on Acinar Cell Bioenergetics
2.4. Effects of MitoQ and DecylTPP on Cell Death, ATP Production and Cell Viability
2.5. Effects of NAC on Cell Death, ATP Production and Cell Viability
3. Discussion
4. Materials and Methods
4.1. Animals and Preparation of Isolated Pancreatic Acinar Cells
4.2. Confocal Microscopy
4.3. Detection of Apoptotic and Necrotic Cell Death Pathways
4.4. Seahorse XF Analysis
4.5. Intracellular ATP Determination
4.6. Lactate Dehydrogenase Assay
4.7. Reagents
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mukherjee, R.; Mareninova, O.A.; Odinokova, I.V.; Huang, W.; Murphy, J.; Chvanov, M.; Javed, M.A.; Wen, L.; Booth, D.M.; Cane, M.C.; et al. Mechanism of mitochondrial permeability transition pore induction and damage in the pancreas: Inhibition prevents acute pancreatitis by protecting production of atp. Gut 2016, 65, 1333–1346. [Google Scholar] [CrossRef]
- Hegyi, P.; Petersen, O.H. The exocrine pancreas: The acinar-ductal tango in physiology and pathophysiology. Rev. Physiol. Biochem. Pharmacol. 2013, 165, 1–30. [Google Scholar] [PubMed]
- Shore, E.R.; Awais, M.; Kershaw, N.M.; Gibson, R.R.; Pandalaneni, S.; Latawiec, D.; Wen, L.; Javed, M.A.; Criddle, D.N.; Berry, N.; et al. Small molecule inhibitors of cyclophilin d to protect mitochondrial function as a potential treatment for acute pancreatitis. J. Med. Chem. 2016, 59, 2596–2611. [Google Scholar] [CrossRef]
- Peery, A.F.; Crockett, S.D.; Murphy, C.C.; Lund, J.L.; Dellon, E.S.; Williams, J.L.; Jensen, E.T.; Shaheen, N.J.; Barritt, A.S.; Lieber, S.R.; et al. Burden and cost of gastrointestinal, liver, and pancreatic diseases in the united states: Update 2018. Gastroenterology 2019, 156, 254–272. [Google Scholar] [CrossRef]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012, CD007176. [Google Scholar] [CrossRef] [PubMed]
- Criddle, D.N.; Gillies, S.; Baumgartner-Wilson, H.K.; Jaffar, M.; Chinje, E.C.; Passmore, S.; Chvanov, M.; Barrow, S.; Gerasimenko, O.V.; Tepikin, A.V.; et al. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J. Biol. Chem. 2006, 281, 40485–40492. [Google Scholar] [CrossRef] [PubMed]
- Siriwardena, A.K.; Mason, J.M.; Balachandra, S.; Bagul, A.; Galloway, S.; Formela, L.; Hardman, J.G.; Jamdar, S. Randomised, double blind, placebo controlled trial of intravenous antioxidant (n-acetylcysteine, selenium, vitamin c) therapy in severe acute pancreatitis. Gut 2007, 56, 1439–1444. [Google Scholar] [CrossRef]
- Armstrong, J.A.; Cash, N.; Soares, P.M.; Souza, M.H.; Sutton, R.; Criddle, D.N. Oxidative stress in acute pancreatitis: Lost in translation? Free Radic. Res. 2013, 47, 917–933. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA 2007, 297, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyurek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re308. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.M.; Murphy, J.A.; Mukherjee, R.; Awais, M.; Neoptolemos, J.P.; Gerasimenko, O.V.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Criddle, D.N. Reactive oxygen species induced by bile acid induce apoptosis and protect against necrosis in pancreatic acinar cells. Gastroenterology 2011, 140, 2116–2125. [Google Scholar] [CrossRef]
- Chvanov, M.; Huang, W.; Jin, T.; Wen, L.; Armstrong, J.; Elliot, V.; Alston, B.; Burdyga, A.; Criddle, D.N.; Sutton, R.; et al. Novel lipophilic probe for detecting near-membrane reactive oxygen species responses and its application for studies of pancreatic acinar cells: Effects of pyocyanin and l-ornithine. Antioxid. Redox Signal. 2015, 22, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.A.; Cash, N.J.; Ouyang, Y.; Morton, J.C.; Chvanov, M.; Latawiec, D.; Awais, M.; Tepikin, A.V.; Sutton, R.; Criddle, D.N. Oxidative stress alters mitochondrial bioenergetics and modifies pancreatic cell death independently of cyclophilin d, resulting in an apoptosis-to-necrosis shift. J. Biol. Chem. 2018, 293, 8032–8047. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Methner, C.; Nadtochiy, S.M.; Logan, A.; Pell, V.R.; Ding, S.; James, A.M.; Cocheme, H.M.; Reinhold, J.; Lilley, K.S.; et al. Cardioprotection by s-nitrosation of a cysteine switch on mitochondrial complex i. Nat. Med. 2013, 19, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant mitoq. Redox Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Dashdorj, A.; Jyothi, K.R.; Lim, S.; Jo, A.; Nguyen, M.N.; Ha, J.; Yoon, K.S.; Kim, H.J.; Park, J.H.; Murphy, M.P.; et al. Mitochondria-targeted antioxidant mitoq ameliorates experimental mouse colitis by suppressing nlrp3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013, 11, 178. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.L.; Desai, R.A.; Bloomfield, P.S.; McIntosh, P.R.; Chapple, K.J.; Linington, C.; Fairless, R.; Diem, R.; Kasti, M.; Murphy, M.P.; et al. Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann. Neurol. 2013, 74, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.K.; Reily, C.; Srivastava, A.; Johnson, M.S.; Ye, Y.; Ulasova, E.; Agarwal, A.; Zinn, K.R.; Murphy, M.P.; Kalyanaraman, B.; et al. Prevention of diabetic nephropathy in ins2(+/)(−)(akitaj) mice by the mitochondria-targeted therapy mitoq. Biochem. J. 2010, 432, 9–19. [Google Scholar] [CrossRef]
- Lowes, D.A.; Webster, N.R.; Murphy, M.P.; Galley, H.F. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br. J. Anaesth. 2013, 110, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant mitoq as a disease-modifying therapy in parkinson’s disease. Mov. Disord. 2010, 25, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Weilert, F.; Orr, D.W.; Keogh, G.F.; Gibson, M.; Lockhart, M.M.; Frampton, C.M.; Taylor, K.M.; Smith, R.A.; Murphy, M.P. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase ii study of hepatitis c patients. Liver. Int. 2010, 30, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic supplementation with a mitochondrial antioxidant (mitoq) improves vascular function in healthy older adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Asin-Cayuela, J.; Manas, A.R.; James, A.M.; Smith, R.A.; Murphy, M.P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004, 571, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Flewelling, R.F.; Hubbell, W.L. The membrane dipole potential in a total membrane potential model. Applications to hydrophobic ion interactions with membranes. Biophys. J. 1986, 49, 541–552. [Google Scholar] [CrossRef]
- Finichiu, P.G.; James, A.M.; Larsen, L.; Smith, R.A.; Murphy, M.P. Mitochondrial accumulation of a lipophilic cation conjugated to an ionisable group depends on membrane potential, ph gradient and pk(a): Implications for the design of mitochondrial probes and therapies. J. Bioenerg. Biomembr. 2013, 45, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Cuenca, S.; Cocheme, H.M.; Logan, A.; Abakumova, I.; Prime, T.A.; Rose, C.; Vidal-Puig, A.; Smith, A.C.; Rubinsztein, D.C.; Fearnley, I.M.; et al. Consequences of long-term oral administration of the mitochondria-targeted antioxidant mitoq to wild-type mice. Free Radic. Biol. Med. 2010, 48, 161–172. [Google Scholar] [CrossRef]
- Reily, C.; Mitchell, T.; Chacko, B.K.; Benavides, G.; Murphy, M.P.; Darley-Usmar, V. Mitochondrially targeted compounds and their impact on cellular bioenergetics. Redox Biol. 2013, 1, 86–93. [Google Scholar] [CrossRef]
- Pokrzywinski, K.L.; Biel, T.G.; Kryndushkin, D.; Rao, V.A. Therapeutic targeting of the mitochondria initiates excessive superoxide production and mitochondrial depolarization causing decreased mtdna integrity. PLoS ONE 2016, 11, e0168283. [Google Scholar] [CrossRef]
- Huang, W.; Cash, N.; Wen, L.; Szatmary, P.; Armstrong, J.; Chvanov, M.; Tepikin, A.; Murphy, M.P.; Sutton, R.; Criddle, D.N. Effects of a mitochondrial-targeted antioxidant mitoquinone (mitoq) in murine experimental acute pancreatitis (ap). Pancreas 2014, 43, 1367. [Google Scholar]
- Gottwald, E.M.; Duss, M.; Bugarski, M.; Haenni, D.; Schuh, C.D.; Landau, E.M.; Hall, A.M. The targeted anti-oxidant mitoq causes mitochondrial swelling and depolarization in kidney tissue. Physiol. Rep. 2018, 6, e13667. [Google Scholar] [CrossRef] [PubMed]
- Dam-Vizi, V.; Chinopoulos, C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends Pharmacol. Sci. 2006, 27, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Indo, H.P.; Hawkins, C.L.; Nakanishi, I.; Matsumoto, K.I.; Matsui, H.; Suenaga, S.; Davies, M.J.; St Clair, D.K.; Ozawa, T.; Majima, H.J. Role of mitochondrial reactive oxygen species in the activation of cellular signals, molecules, and function. Handb. Exp. Pharmacol. 2017, 240, 439–456. [Google Scholar] [PubMed]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Brand, M.D.; Affourtit, C.; Esteves, T.C.; Green, K.; Lambert, A.J.; Miwa, S.; Pakay, J.L.; Parker, N. Mitochondrial superoxide: Production, biological effects, and activation of uncoupling proteins. Free Radic. Biol. Med. 2004, 37, 755–767. [Google Scholar] [CrossRef]
- Arsenijevic, D.; Onuma, H.; Pecqueur, C.; Raimbault, S.; Manning, B.S.; Miroux, B.; Couplan, E.; Alves-Guerra, M.C.; Goubern, M.; Surwit, R.; et al. Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat. Genet. 2000, 26, 435–439. [Google Scholar] [CrossRef]
- de Bilbao, F.; Arsenijevic, D.; Vallet, P.; Hjelle, O.P.; Ottersen, O.P.; Bouras, C.; Raffin, Y.; Abou, K.; Langhans, W.; Collins, S.; et al. Resistance to cerebral ischemic injury in ucp2 knockout mice: Evidence for a role of ucp2 as a regulator of mitochondrial glutathione levels. J. Neurochem. 2004, 89, 1283–1292. [Google Scholar] [CrossRef]
- Segersvard, R.; Rippe, C.; Duplantier, M.; Herrington, M.K.; Isaksson, B.; Adrian, T.E.; Erlanson-Albertsson, C.; Permert, J. Mrna for pancreatic uncoupling protein 2 increases in two models of acute experimental pancreatitis in rats and mice. Cell Tissue Res. 2005, 320, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Leo, S.; Szabadkai, G.; Rizzuto, R. The mitochondrial antioxidants mitoe(2) and mitoq(10) increase mitochondrial ca(2+) load upon cell stimulation by inhibiting ca(2+) efflux from the organelle. Ann. N. Y. Acad. Sci. 2008, 1147, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Voronina, S.; Sukhomlin, T.; Johnson, P.R.; Erdemli, G.; Petersen, O.H.; Tepikin, A. Correlation of nadh and ca2+ signals in mouse pancreatic acinar cells. J. Physiol. 2002, 539, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.A.; Criddle, D.N.; Sherwood, M.; Chvanov, M.; Mukherjee, R.; McLaughlin, E.; Booth, D.; Gerasimenko, J.V.; Raraty, M.G.; Ghaneh, P.; et al. Direct activation of cytosolic ca2+ signaling and enzyme secretion by cholecystokinin in human pancreatic acinar cells. Gastroenterology 2008, 135, 632–641. [Google Scholar] [CrossRef]
- Lu, L.Y.; Ou, N.; Lu, Q.B. Antioxidant induces DNA damage, cell death and mutagenicity in human lung and skin normal cells. Sci. Rep. 2013, 3, 3169. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Cash, N.; Wen, L.; Szatmary, P.; Mukherjee, R.; Armstrong, J.; Chvanov, M.; Tepikin, A.V.; Murphy, M.P.; Sutton, R.; et al. Effects of the mitochondria-targeted antioxidant mitoquinone in murine acute pancreatitis. Mediat. Inflamm. 2015, 2015, 901780. [Google Scholar] [CrossRef] [PubMed]
- Sansbury, B.E.; Jones, S.P.; Riggs, D.W.; Darley-Usmar, V.M.; Hill, B.G. Bioenergetic function in cardiovascular cells: The importance of the reserve capacity and its biological regulation. Chem. Biol. Interact. 2011, 191, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Yadava, N.; Nicholls, D.G. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex i with rotenone. J. Neurosci. 2007, 27, 7310–7317. [Google Scholar] [CrossRef] [PubMed]
- Criddle, D.N.; Murphy, J.; Fistetto, G.; Barrow, S.; Tepikin, A.V.; Neoptolemos, J.P.; Sutton, R.; Petersen, O.H. Fatty acid ethyl esters cause pancreatic calcium toxicity via inositol trisphosphate receptors and loss of atp synthesis. Gastroenterology 2006, 130, 781–793. [Google Scholar] [CrossRef]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar] [CrossRef]
- Beger, H.G.; Rau, B.M. Severe acute pancreatitis: Clinical course and management. World J. Gastroenterol. 2007, 13, 5043–5051. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.M.; Saluja, A.K.; Sengupta, A.; Saluja, M.; Steer, M.L. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am. J. Physiol. 1995, 269, C1295–C1304. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Wallig, M.A.; Hofbauer, B.; Lee, H.S.; Frossard, J.L.; Steer, M.L.; Saluja, A.K. Induction of apoptosis in pancreatic acinar cells reduces the severity of acute pancreatitis. Biochem. Biophys. Res. Commun. 1998, 246, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Schoener, B.; Oshino, R.; Itshak, F.; Nakase, Y. Oxidation-reduction ratio studies of mitochondria in freeze-trapped samples. Nadh and flavoprotein fluorescence signals. J. Biol. Chem. 1979, 254, 4764–4771. [Google Scholar] [PubMed]
- Ostrander, J.H.; McMahon, C.M.; Lem, S.; Millon, S.R.; Brown, J.Q.; Seewaldt, V.L.; Ramanujam, N. Optical redox ratio differentiates breast cancer cell lines based on estrogen receptor status. Cancer Res. 2010, 70, 4759–4766. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Goncalves, R.L.S.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta 2015, 1847, 171–181. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armstrong, J.A.; Cash, N.J.; Morton, J.C.; Tepikin, A.V.; Sutton, R.; Criddle, D.N. Mitochondrial Targeting of Antioxidants Alters Pancreatic Acinar Cell Bioenergetics and Determines Cell Fate. Int. J. Mol. Sci. 2019, 20, 1700. https://doi.org/10.3390/ijms20071700
Armstrong JA, Cash NJ, Morton JC, Tepikin AV, Sutton R, Criddle DN. Mitochondrial Targeting of Antioxidants Alters Pancreatic Acinar Cell Bioenergetics and Determines Cell Fate. International Journal of Molecular Sciences. 2019; 20(7):1700. https://doi.org/10.3390/ijms20071700
Chicago/Turabian StyleArmstrong, Jane A., Nicole J. Cash, Jack C. Morton, Alexei V. Tepikin, Robert Sutton, and David N. Criddle. 2019. "Mitochondrial Targeting of Antioxidants Alters Pancreatic Acinar Cell Bioenergetics and Determines Cell Fate" International Journal of Molecular Sciences 20, no. 7: 1700. https://doi.org/10.3390/ijms20071700