Functional Characterization of Colon Cancer-Associated Mutations in ADAM17: Modifications in the Pro-Domain Interfere with Trafficking and Maturation

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Mutations in ADAM17 Are Associated with Colon Cancer
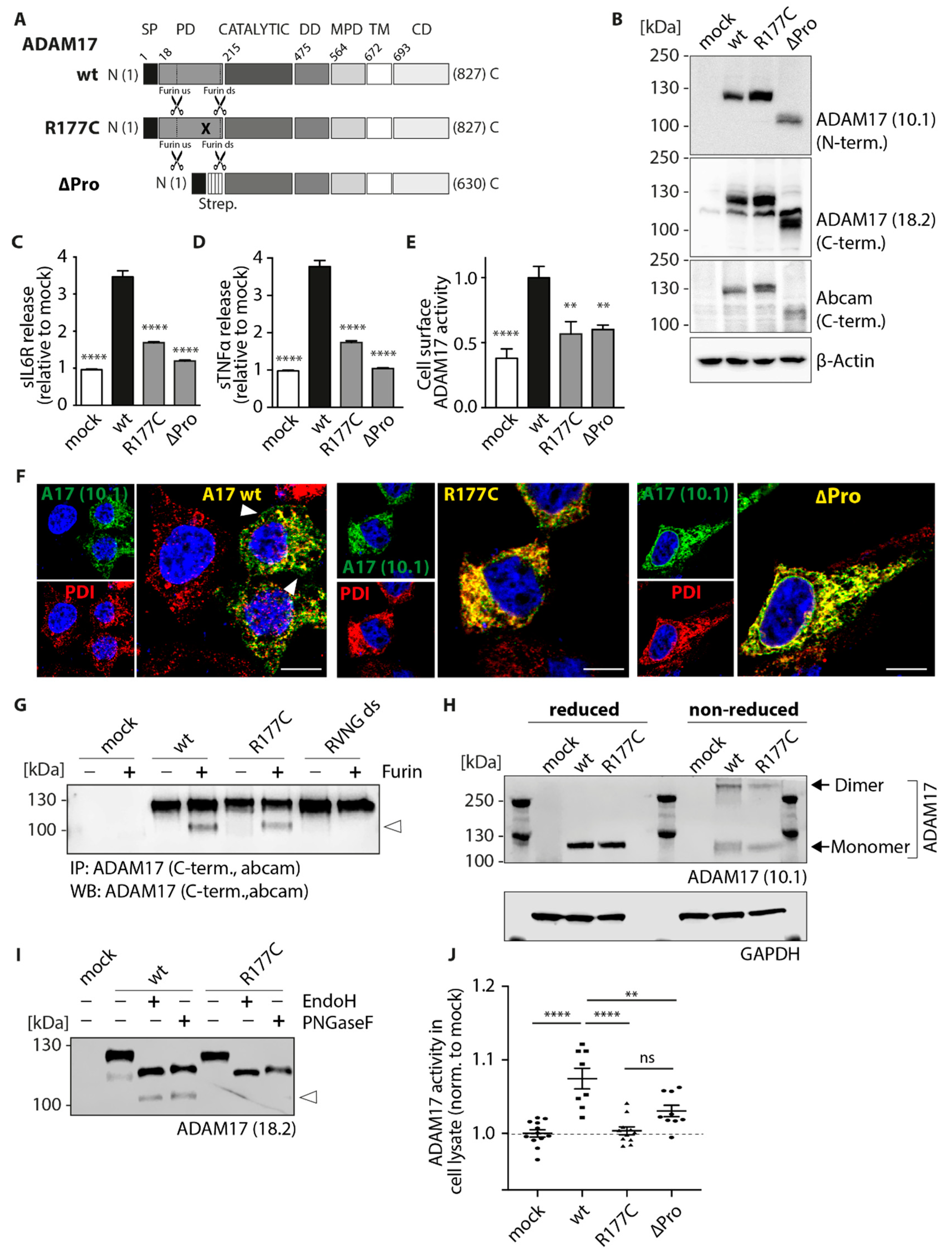
2.2. Colon Cancer-Associated ADAM17 Variants Differ in Their Proteolytic Activity
2.3. Cellular Localization Is Altered in Colon Cancer-Associated ADAM17 Variants
2.4. A Colon Cancer-Associated ADAM17 Mutation within the Pro-Domain Is Catalytically as Inactive as an ADAM17 Variant Lacking the Entire Pro-Domain
3. Discussion
4. Material and Methods
4.1. Databases
4.2. cDNA Constructs and Cloning
4.3. Cell Culture
4.4. Transfection
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Flow Cytometry
4.7. Immunofluorescence Analysis
4.8. Western Blot Analysis
4.9. Deglycosylation Analysis
4.10. Immunoprecipitation and Furin Cleavage Assay
4.11. Live Cell Surface ADAM17 Activity Assay
4.12. Cell Lysate ADAM17 Activity Assay
4.13. Structural Analysis
4.14. Data Analysis and Statistics
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aa | amino acid |
| ADAM17 | A Disintegrin and Metalloproteinase 17 |
| CD | cytoplasmic domain |
| COSMIC | Catalogue of Somatic Mutations in Cancer |
| dKO | double knock-out |
| ds | downstream |
| EGF-R | epidermal growth factor receptor |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| ER | endoplasmic reticulum |
| FGF-2 | fibroblast growth factor-2 |
| FSC | forward scatter |
| HEK | human embryonic kidney cells |
| ICGC | International Cancer Genome Consortium |
| IL-6R | interleukin-6 receptor |
| IntOGen | Integrative Onco Genomics |
| kDa | kilo Dalton |
| MPD | membrane-proximal domain |
| PD | pro-domain |
| PDI | protein disulfide isomerase |
| PMA | phorbol 12-myristate 13-acetate |
| sTNFα | soluble tumor necrosis factor alpha |
| TCGA | The Cancer Genome Atlas Program |
| us | upstream |
| VEGF | vascular endothelial growth factor |
| wt | wildtype |
References
- Siegel, R.; Desantis, C.; Jemal, A. Colorectal cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 104–117. [Google Scholar]
- Rustgi, A.K. The genetics of hereditary colon cancer. Genes Dev. 2007, 21, 2525–2538. [Google Scholar] [CrossRef] [Green Version]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114. [Google Scholar] [CrossRef] [PubMed]
- Sibilia, M.; Kroismayr, R.; Lichtenberger, B.M.; Natarajan, A.; Hecking, M.; Holcmann, M. The epidermal growth factor receptor: From development to tumorigenesis. Differ. Res. Biol. Divers. 2007, 75, 770–787. [Google Scholar] [CrossRef]
- Egger, B.; Buchler, M.W.; Lakshmanan, J.; Moore, P.; Eysselein, V.E. Mice harboring a defective epidermal growth factor receptor (waved-2) have an increased susceptibility to acute dextran sulfate-induced colitis. Scand. J. Gastroenterol. 2000, 35, 1181–1187. [Google Scholar] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Jin, S.L.; Milla, M.E.; Bickett, D.M.; Burkhart, W.; Carter, H.L.; Chen, W.J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef]
- Blobel, C.P. ADAMs: Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Adam, N.; Sina, C.; Rosenstiel, P.; Lehmann-Koch, J.; Schirmacher, P.; Hartmann, D.; Cichy, J.; Gavrilova, O.; Schreiber, S.; et al. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 2010, 207, 1617–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaydon, D.C.; Biancheri, P.; Di, W.L.; Plagnol, V.; Cabral, R.M.; Brooke, M.A.; van Heel, D.A.; Ruschendorf, F.; Toynbee, M.; Walne, A.; et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 2011, 365, 1502–1508. [Google Scholar] [CrossRef]
- Peschon, J.J.; Slack, J.L.; Reddy, P.; Stocking, K.L.; Sunnarborg, S.W.; Lee, D.C.; Russell, W.E.; Castner, B.J.; Johnson, R.S.; Fitzner, J.N.; et al. An essential role for ectodomain shedding in mammalian development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef]
- Schmidt, S.; Schumacher, N.; Schwarz, J.; Tangermann, S.; Kenner, L.; Schlederer, M.; Sibilia, M.; Linder, M.; Altendorf-Hofmann, A.; Knösel, T.; et al. ADAM17 is required for EGF-R–induced intestinal tumors via IL-6 trans-signaling. J. Exp. Med. 2018, 215, 1205–1225. [Google Scholar] [CrossRef] [PubMed]
- Jostock, T.; Mullberg, J.; Ozbek, S.; Atreya, R.; Blinn, G.; Voltz, N.; Fischer, M.; Neurath, M.F.; Rose-John, S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 2001, 268, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, E.; Borrelli, F.; Orlando, P.; Romano, B.; Monti, M.; Morbidelli, L.; Aviello, G.; Imperatore, R.; Capasso, R.; Piscitelli, F.; et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res. 2017, 119, 227–236. [Google Scholar] [CrossRef]
- Yu, W.; Ma, Y.; Shankar, S.; Srivastava, R.K. SATB2/beta-catenin/TCF-LEF pathway induces cellular transformation by generating cancer stem cells in colorectal cancer. Sci. Rep. 2017, 7, 10939. [Google Scholar] [CrossRef]
- Wei, L.H.; Chou, C.H.; Chen, M.W.; Rose-John, S.; Kuo, M.L.; Chen, S.U.; Yang, Y.S. The role of IL-6 trans-signaling in vascular leakage: Implications for ovarian hyperstimulation syndrome in a murine model. J. Clin. Endocrinol. Metab. 2013, 98, E472–E484. [Google Scholar] [CrossRef]
- Leonard, J.D.; Lin, F.; Milla, M.E. Chaperone-like properties of the prodomain of TNFalpha-converting enzyme (TACE) and the functional role of its cysteine switch. Biochem. J. 2005, 387, 797–805. [Google Scholar] [CrossRef]
- Wong, E.; Maretzky, T.; Peleg, Y.; Blobel, C.P.; Sagi, I. The Functional Maturation of A Disintegrin and Metalloproteinase (ADAM) 9, 10, and 17 Requires Processing at a Newly Identified Proprotein Convertase (PC) Cleavage Site. J. Biol. Chem. 2015, 290, 12135–12146. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cohen, T.; Romi, E.; Levin, M.; Peleg, Y.; Arad, U.; Yaron, A.; Milla, M.E.; Sagi, I. Harnessing the natural inhibitory domain to control TNFalpha Converting Enzyme (TACE) activity in vivo. Sci. Rep. 2016, 6, 35598. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Minond, D. Recent Advances in ADAM17 Research: A Promising Target for Cancer and Inflammation. Mediat. Inflamm. 2017, 2017, 9673537. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Lokau, J.; Dusterhoft, S.; Trad, A.; Garbers, C.; Scheller, J.; Rose-John, S.; Grotzinger, J. The membrane-proximal domain of A Disintegrin and Metalloprotease 17 (ADAM17) is responsible for recognition of the interleukin-6 receptor and interleukin-1 receptor II. FEBS Lett. 2012, 586, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.; Slack, J.L.; Davis, R.; Cerretti, D.P.; Kozlosky, C.J.; Blanton, R.A.; Shows, D.; Peschon, J.J.; Black, R.A. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem. 2000, 275, 14608–14614. [Google Scholar] [CrossRef]
- Doedens, J.R.; Mahimkar, R.M.; Black, R.A. TACE/ADAM-17 enzymatic activity is increased in response to cellular stimulation. Biochem. Biophys. Res. Commun. 2003, 308, 331–338. [Google Scholar] [CrossRef]
- Le Gall, S.M.; Maretzky, T.; Issuree, P.D.; Niu, X.D.; Reiss, K.; Saftig, P.; Khokha, R.; Lundell, D.; Blobel, C.P. ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci. 2010, 123, 3913–3922. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Evers, A.; Zhou, W.; Swendeman, S.L.; Wong, P.M.; Rafii, S.; Reiss, K.; Blobel, C.P. Migration of growth factor-stimulated epithelial and endothelial cells depends on EGFR transactivation by ADAM17. Nat. Commun. 2011, 2, 229. [Google Scholar] [CrossRef]
- Schwarz, J.; Broder, C.; Helmstetter, A.; Schmidt, S.; Yan, I.; Muller, M.; Schmidt-Arras, D.; Becker-Pauly, C.; Koch-Nolte, F.; Mittrucker, H.W.; et al. Short-term TNFalpha shedding is independent of cytoplasmic phosphorylation or furin cleavage of ADAM17. Biochim. Biophys. Acta 2013, 1833, 3355–3367. [Google Scholar] [CrossRef]
- Cabron, A.S.; El Azzouzi, K.; Boss, M.; Arnold, P.; Schwarz, J.; Rosas, M.; Dobert, J.P.; Pavlenko, E.; Schumacher, N.; Renne, T.; et al. Structural and Functional Analyses of the Shedding Protease ADAM17 in HoxB8-Immortalized Macrophages and Dendritic-like Cells. J. Immunol. 2018, 201, 3106–3118. [Google Scholar] [CrossRef] [PubMed]
- Mullberg, J.; Schooltink, H.; Stoyan, T.; Gunther, M.; Graeve, L.; Buse, G.; Mackiewicz, A.; Heinrich, P.C.; Rose-John, S. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993, 23, 473–480. [Google Scholar] [CrossRef]
- Soond, S.M.; Everson, B.; Riches, D.W.; Murphy, G. ERK-mediated phosphorylation of Thr735 in TNFalpha-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 2005, 118, 2371–2380. [Google Scholar] [CrossRef] [PubMed]
- Killock, D.J.; Ivetic, A. The cytoplasmic domains of TNFalpha-converting enzyme (TACE/ADAM17) and L-selectin are regulated differently by p38 MAPK and PKC to promote ectodomain shedding. Biochem. J. 2010, 428, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Derynck, R. Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 2010, 37, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Liu, J.; Sakaki-Yumoto, M.; Derynck, R. TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 2012, 5, ra34. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Turck, C.W.; Derynck, R. Characterization of growth factor-induced serine phosphorylation of tumor necrosis factor-alpha converting enzyme and of an alternatively translated polypeptide. J. Biol. Chem. 2003, 278, 18617–18627. [Google Scholar] [CrossRef]
- Peiretti, F.; Canault, M.; Deprez-Beauclair, P.; Berthet, V.; Bonardo, B.; Juhan-Vague, I.; Nalbone, G. Intracellular maturation and transport of tumor necrosis factor alpha converting enzyme. Exp. Cell Res. 2003, 285, 278–285. [Google Scholar] [CrossRef]
- Mustafi, R.; Dougherty, U.; Mustafi, D.; Ayaloglu-Butun, F.; Fletcher, M.; Adhikari, S.; Sadiq, F.; Meckel, K.; Haider, H.I.; Khalil, A.; et al. ADAM17 is a Tumor Promoter and Therapeutic Target in Western Diet-associated Colon Cancer. Clin. Cancer Res. 2017, 23, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Spano, J.P.; Lagorce, C.; Atlan, D.; Milano, G.; Domont, J.; Benamouzig, R.; Attar, A.; Benichou, J.; Martin, A.; Morere, J.F.; et al. Impact of EGFR expression on colorectal cancer patient prognosis and survival. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2005, 16, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Haraldsdottir, S.; Bekaii-Saab, T. Integrating anti-EGFR therapies in metastatic colorectal cancer. J. Gastrointest. Oncol. 2013, 4, 285–298. [Google Scholar]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schuler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef]
- Srivatsa, S.; Paul, M.C.; Cardone, C.; Holcmann, M.; Amberg, N.; Pathria, P.; Diamanti, M.A.; Linder, M.; Timelthaler, G.; Dienes, H.P.; et al. EGFR in Tumor-Associated Myeloid Cells Promotes Development of Colorectal Cancer in Mice and Associates With Outcomes of Patients. Gastroenterology 2017, 153, 178–190.e10. [Google Scholar] [CrossRef]
- Gonzales, P.E.; Solomon, A.; Miller, A.B.; Leesnitzer, M.A.; Sagi, I.; Milla, M.E. Inhibition of the tumor necrosis factor-alpha-converting enzyme by its pro domain. J. Biol. Chem. 2004, 279, 31638–31645. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yan, Y.; Huang, W.; Yang, Y.; Wang, H.; Chang, L. The regulation of TACE catalytic function by its prodomain. Mol. Biol. Rep. 2009, 36, 641–651. [Google Scholar] [CrossRef]
- Saad, M.I.; Alhayyani, S.; McLeod, L.; Yu, L.; Alanazi, M.; Deswaerte, V.; Tang, K.; Jarde, T.; Smith, J.A.; Prodanovic, Z.; et al. ADAM17 selectively activates the IL-6 trans-signaling/ERK MAPK axis in KRAS-addicted lung cancer. EMBO Mol. Med. 2019, 11, e9976. [Google Scholar] [CrossRef]
- Milla, M.E.; Leesnitzer, M.A.; Moss, M.L.; Clay, W.C.; Carter, H.L.; Miller, A.B.; Su, J.L.; Lambert, M.H.; Willard, D.H.; Sheeley, D.M.; et al. Specific sequence elements are required for the expression of functional tumor necrosis factor-alpha-converting enzyme (TACE). J. Biol. Chem. 1999, 274, 30563–30570. [Google Scholar] [CrossRef]
- Chavaroche, A.; Cudic, M.; Giulianotti, M.; Houghten, R.A.; Fields, G.B.; Minond, D. Glycosylation of a disintegrin and metalloprotease 17 affects its activity and inhibition. Anal. Biochem. 2014, 449, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. Lessons from hereditary colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef]
- McFarland, C.D.; Yaglom, J.A.; Wojtkowiak, J.W.; Scott, J.G.; Morse, D.L.; Sherman, M.Y.; Mirny, L.A. The Damaging Effect of Passenger Mutations on Cancer Progression. Cancer Res. 2017, 77, 4763–4772. [Google Scholar] [CrossRef]
- Pon, J.R.; Marra, M.A. Driver and passenger mutations in cancer. Annu. Rev. Pathol. 2015, 10, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Riethmueller, S.; Ehlers, J.C.; Lokau, J.; Dusterhoft, S.; Knittler, K.; Dombrowsky, G.; Grotzinger, J.; Rabe, B.; Rose-John, S.; Garbers, C. Cleavage Site Localization Differentially Controls Interleukin-6 Receptor Proteolysis by ADAM10 and ADAM17. Sci. Rep. 2016, 6, 25550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seegar, T.C.M.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the alpha-Secretase ADAM10. Cell 2017, 171, 1638–1648.7. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlenko, E.; Cabron, A.-S.; Arnold, P.; Dobert, J.P.; Rose-John, S.; Zunke, F. Functional Characterization of Colon Cancer-Associated Mutations in ADAM17: Modifications in the Pro-Domain Interfere with Trafficking and Maturation. Int. J. Mol. Sci. 2019, 20, 2198. https://doi.org/10.3390/ijms20092198
Pavlenko E, Cabron A-S, Arnold P, Dobert JP, Rose-John S, Zunke F. Functional Characterization of Colon Cancer-Associated Mutations in ADAM17: Modifications in the Pro-Domain Interfere with Trafficking and Maturation. International Journal of Molecular Sciences. 2019; 20(9):2198. https://doi.org/10.3390/ijms20092198
Chicago/Turabian StylePavlenko, Egor, Anne-Sophie Cabron, Philipp Arnold, Jan Philipp Dobert, Stefan Rose-John, and Friederike Zunke. 2019. "Functional Characterization of Colon Cancer-Associated Mutations in ADAM17: Modifications in the Pro-Domain Interfere with Trafficking and Maturation" International Journal of Molecular Sciences 20, no. 9: 2198. https://doi.org/10.3390/ijms20092198