Chemotherapy-Induced Peripheral Neuropathy and Changes in Cytoskeleton


School of Medicine and Surgery, Experimental Neurology Unit and Milan Center for Neuroscience, University of Milano-Bicocca, via Cadore 48, 20900 Monza, MB, Italy
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2019, 20(9), 2287; https://doi.org/10.3390/ijms20092287
Submission received: 25 February 2019
/
Revised: 6 May 2019
/
Accepted: 7 May 2019
/
Published: 9 May 2019
(This article belongs to the Special Issue Molecular and Cellular Mechanisms of Neurotoxicity)
Abstract
:Despite the different antineoplastic mechanisms of action, peripheral neurotoxicity induced by all chemotherapy drugs (anti-tubulin agents, platinum compounds, proteasome inhibitors, thalidomide) is associated with neuron morphological changes ascribable to cytoskeleton modifications. The “dying back” degeneration of distal terminals (sensory nerves) of dorsal root ganglia sensory neurons, observed in animal models, in in vitro cultures and biopsies of patients is the most evident hallmark of the perturbation of the cytoskeleton. On the other hand, in highly polarized cells like neurons, the cytoskeleton carries out its role not only in axons but also has a fundamental role in dendrite plasticity and in the organization of soma. In the literature, there are many studies focused on the antineoplastic-induced alteration of microtubule organization (and consequently, fast axonal transport defects) while very few studies have investigated the effect of the different classes of drugs on microfilaments, intermediate filaments and associated proteins. Therefore, in this review, we will focus on: (1) Highlighting the fundamental role of the crosstalk among the three filamentous subsystems and (2) investigating pivotal cytoskeleton-associated proteins.
1. Chemotherapy-Induced Peripheral Neuropathy (CIPN)
Chemotherapy-induced peripheral neuropathy (CIPN) is a debilitating and dose-limiting side effect manifested mainly by a sensory, length-dependent process, that results from a drug cumulative dose. CIPN symptoms can be sufficiently severe to require a reduction in drug dosage or discontinuation of treatment [1], causing a significant hindrance to favorable outcomes and long-term patient quality of life. The development of more efficient antitumor therapy has led to a steady increase in the survival rates of patients, with a subset of them who experience chronic CIPN symptoms that continue even after the end of the therapy. As a result, a focus on late toxicity of commonly used antineoplastic drugs has emerged.
CIPN is associated with many drugs that differ both in their antineoplastic action and in the proposed neurotoxic mechanism.
CIPN has been reported in patients treated with platinum-based drugs, including cisplatin and oxaliplatin, microtubule-targeting agents (MTA) [2,3] such as taxanes (paclitaxel and docetaxel), vinca alkaloids (particularly vincristine and vinblastine), epothilones and eribulin, and also proteasome inhibitors (bortezomib) and immunomodulatory drugs (thalidomide).
Although the specific anticancer effect of all classes of drugs is well known, their molecular and cellular impact on the peripheral nervous system is not completely clear. While some classes of antineoplastic drugs have potentially the antiproliferative mechanism strictly linked to their neurotoxicity action (i.e., anti-tubulin compounds), others, for example, the platinum-based drugs, show different neurotoxic effects unrelated to their antineoplastic activity (Table 1).
Below are the classical antitumor mechanisms of action of the different classes of antineoplastic drugs.
- Epothilones exert their effect by stabilizing microtubules which leads to apoptosis in cancer cells [18].
- Eribulin is known to bind at the plus (+) ends of the microtubule, inducing an increase of microtubule depolymerization [16].
- Proteasome inhibitors exert their antiproliferative action by inhibiting the proteasome—the primary intracellular protein degradation machinery—which prevents proteolytic cleavage of intracellular proteins and results in protein aggregate accumulation in tumor cells, leading to cell cycle arrest and apoptosis [22,23].
- Thalidomide mechanism of action is poorly understood but immunomodulation and antiangiogenic effects appear to be involved in its antiproliferative activity, as well as down-regulation of tumor necrosis factor alpha (TNFα) [24].
CIPN in cancer patients generally emerges with various sensory symptoms including paresthesia and dysesthesia, heat/cold hyperalgesia and tingling, which can degenerate into severe sensory and motor damages. Moreover, painful symptoms, such as burning sensation and/or stabbing pain, can occur [25]. Clinically, these manifestations develop in a glove and stocking distribution due to preferential neurotoxic effect on longer axons. Depending on the anti-tumor treatment, the neuropathic symptoms will differ in degree of severity, clinical features and recovery [26]. CIPN current clinical strategies primarily aim to relieve symptoms, focusing on pain.
Due to the lack of knowledge of the pathogenic mechanisms of CIPN, preventive or symptomatic treatments are usually ineffective and neuroprotective agents to prevent or manage CIPN are inadequate. Many efforts have been made to develop strategies of prevention and treatment of CIPN for the recovery of the patient’s condition and quality of life, but at present, there are no established therapeutic strategies to prevent these adverse events [1,27]. Many groups are investigating various hypotheses about the multifactorial pathophysiology underlying CIPN [25,28,29]. In fact, it was reported that CIPN onset involves damage to both glial and neuronal cells and related oxidative stress-mediated damage to cells [11]. In addition, cytokine activation, regulation of intracellular signaling pathways, changes of neuronal ion channel responses, impairment of axonal trafficking, and immune system activation were also investigated as putative players involved in CIPN [9] (Table 1). In any case, one of the most frequently investigated mechanisms for the pathogenesis of CIPN involves mitochondrial damage in glia and neurons [11]. Therefore, protecting mitochondrial impairment has been proposed as a promising therapeutic approach to improve clinical management of this side effect. Other studies have also shown that impairment of Ca2+ homeostasis—which ultimately leads to apoptosis and neuroinflammation—as possible targets for CIPN treatment [30]. Furthermore, genetic influences (including single polymorphisms) are associated with the risk of developing CIPN, although it is unclear how many actually contribute mechanistically to CIPN [8].
2. CIPN Models
Considering the high recurrence of CIPN and its limiting effect on chemotherapeutic treatment, numerous reliable animal models have been adopted to investigate it.
Chemotherapy drugs are typically administered to mice and rats through gavage, intravenous or intraperitoneal injection. Endpoint parameters to measure CIPN are the intraepidermal unmyelinated axon density [31,32], behavioral parameters, and morphological and morphometrical analysis of neuronal soma and nerve fibers [33,34,35]. The main behavioral tests to evaluate mechanical allodynia (i.e., hypersensitivity to normally innocuous stimuli), thermal algesia and motor performance are automatic or manual von Frey, plantar, tail-flick, cold plate and rota-rod [36]. In addition to the behavioral aspect, animal models give important information at the electrophysiological level in order to characterize neurotoxicity. Therefore, nerve conduction velocity and nerve action potential are usually analyzed in the caudal and digital nerves [37]. Furthermore, the histological characterization of neuronal morphology and quantity, as well as nerve morphometry, are essential to describe axonal or myelin damage, axonal density or the distribution of axonal diameters of myelinated fibers [36]. All of these methods allow investigation of the peripheral nervous system structure changes involved in neuronopathy, axonopathy or myelinopathy [38].
Sensory neurons and their axons, but also satellite cells, are very susceptible to damage induced by antitumor treatment because of deficiency in the blood-nerve barrier, which allows easy permeation of antitumor drugs [39]. For this reason, in addition to animal models, numerous in vitro studies have been carried out, mainly on dorsal root ganglia (DRG) cultures, in order to investigate the molecular mechanisms of neurotoxicity. In fact, the DRG is the primary target of antitumor compounds causing a predominantly sensory pathology in patients treated with chemotherapy [40].
Different DRG-based in vitro models have been proposed in the literature: Whole DRG organotypic cultures of mice or rat embryos [41]; primary culture of (embryonal or adult) mice or rat DRG sensory neurons further treated with antineoplastic drugs [41,42]; PC12 pheochromocytoma cells [43]. In these models, the quantification of neurite outgrowth, as a functional endpoint, was used to reflect specific chemotherapy-induced neurotoxicity [44]. This parameter can be quantified with different techniques. The first method consists of a manual measure of the length of the longest neurite in each DRG by the ImageJ program [41]. A semi-automatic method, the NeuronJ plugin for ImageJ, has been developed [45]. In addition, new automatic methods have emerged based on image processing software, like ImagePro [46]. Wu and Bradshaw have used a standard phase contrast microscope to study neurotoxicity in PC12 cells [47]; Bilsland et al. have quantified, by a semi-quantitative analysis, the area occupied by neurites emanating from chick DRG explants [48]; and Popova and Jacobsson have used a fluorescent microplate assay to study βIII-tubulin immunoreactivity in P19-derived neurons [49]. Using these models, several neuroprotective substances have been tested, such as nerve growth factor (NGF) [50], as well ciliary neurotrophic factors (CNTF), brain-derived neurotrophic factors [41] or basic fibroblast growth factors [51].
In addition, ex vivo DRG cultures from adult mice treated with neurotoxic antineoplastic drugs have been used to study the mechanism underlying CIPN. This model allows preservation of a heterogeneous system, containing multiple cell types residing in a DRG, including the neurons, resident macrophages and glial cells [52]. The multiparametric morphology-centered rat DRG assay developed by Guo et al., dynamically measuring the chemotherapy-induced morphological alterations in the subcellular structures of neurons and non-neuron cells, appears to be a good model for highlighting the effect of an antineoplastic drug [53].
Lastly, human induced pluripotent stem cell (iPSC) derived peripheral-like neurons have often been used to predict peripheral neurotoxicants by morphological characterization and viability using high-content imaging analysis [54,55,56,57]. This approach will allow both mechanistic studies and screening of potential neuroprotective substances to prevent CIPN. The human iPSC-derived neuron represents a more favorable model than dissociated rodent neuronal culture for its high translatability to humans, due to a similar neuronal structure, which are hardly available otherwise. Therefore, in neurons derived from the iPSC model, it was demonstrated that antitumor drugs can impact neurite length independently from general cytotoxicity [54].
Moreover, some non-mammalian models have also been used to understand the mechanisms behind CIPN, such as Zebrafish or Drosophila models, but they are less usually studied [12,58,59,60]. The study of these models permits further understanding of numerous biological processes, due to similarities in many mammalian genes, rapid development with short life cycles, ready availability and easy manipulation of the genetic system. However, they cannot reproduce the complexity of the human system.
3. Neuronal Cytoskeleton
Successful function and development of the nervous system depend on the correct function of the cytoskeletal machinery. Cell migration, proliferation, neuron polarization and synapse network establishment are complex processes coordinated by the structural organization and dynamic remodeling of the neuronal cytoskeleton (CTK) (Figure 1). In humans, there are numerous disorders resulting from the alteration or damage of the myriad of players involved in the neuronal CTK machinery, both in the central and peripheral nervous systems. These causes vary from purely genetic to the pharmacological [61,62,63].
Three main polymers constitute the backbone of the elaborated and, to some extent, unknown neuronal CTK: The microfilaments, the intermediate filaments and the microtubules. However, in order to achieve proper performance, the CTK needs to associate and interact with a high number of other proteins [64].
Microtubules (MTs) are an array of α and β-tubulin heterodimers stuck together in a head-to-tail fashion, which laterally form a rather stiff, hollow tubular structure of about 25 nm diameter [65]. In neurons, bundles of MTs are kept together and stabilized by structural microtubule-associated proteins (MAP) crosslinks: Tau in axons and MAP2 in dendrites and soma. MTs are polarized structures with a plus-end and a minus-end. In axons, the plus ends are oriented towards the tip of the axon but in dendrites, MTs present a mixed polarity [66]. Another class of MAPs, called microtubule plus-end-tracking proteins (+TIPS), regulate microtubule dynamics: Polymerization, depolymerization and catastrophe phenomena. In addition, the +TIPS EB1 and EB3 recruit additional players involved in the regulation of the local axonal transport [67].
In fact, given that neurons are highly polarized cells, dendritic and axonal transport machinery are essential for the delivery of organelles, proteins and RNA to the distal ends (anterograde transport) in order to keep structural turnover and synaptic activity. This is also essential for the retrograde transport of aging components towards the cell body for being recycled and degraded [68,69]. In human adults for example, the length of peripheral fibers can reach 1 m, and thus the transport along axons, from the cellular body to neuromuscular junctions (or the other way around from receptors), requires a highly organized microtubule network.
The kinesin protein family is a large group of motor proteins highly expressed in neurons, where they move along microtubules in order to deliver their cargo toward the plus-end tip of the axons. In particular, motor protein kinesin-1 acts selectively, transporting cargoes in axons. This is the case even in dendrites, where it is possible to find plus-end tip microtubules distally oriented. On the other hand, cytoplasm dynein motor protein is the principal molecule responsible for retrograde transport in axons, toward the MT minus-end tip. In dendrites, instead, the unique existence of distally oriented minus-ends consents in a selective way to the minus-end-directed motor dynein to transport cargo towards dendrites tips [65,70].
One of the key mechanisms that regulate axonal transport and MT dynamics in neurons is the post-translational modification of tubulin, including as polyglutamylation, phosphorylation, polyglycylation and detyrosination. Acetylation of α-tubulin at lysine-40 by the enzyme α-tubulin acetyltransferase 1 (aTAT1) is the most studied of these modifications and is the only one that takes place in the lumen of the MT. This specific modification is present in almost all cell types and is very well conserved. Moreover, MT acetylation at lysine-40 seems to be a modification that characterizes stable long-lived MTs [71]. In fact, the absence of lysine-40 acetylation in MTs has been shown in Caenorhabditis elegans to lead to neuronal degeneration [72]. It has also been reported that increased acetylation could recruit and improve the docking of motor proteins to MT [73]. In addition, recent studies demonstrate that MT acetylation at lysine-40 plays an important role in mechanosensation in mammals, where sensory peripheral neurons axonal MT are highly acetylated. This modification seems to regulate the membrane stiffness of sensory neurons, tuning the ideal grade of elasticity for the best mechanical touch and pain detection [74].
Considering that the state of MT acetylation depends on the activity of deacetylase enzymes, some papers have investigated histone deacetylase 6 (HDAC6), an atypical cytoplasmic histone deacetylase. Unlike conventional histone deacetylase, HDAC is mostly located in the cytoplasm and has two catalytic sites. These features confer to HDAC6 the ability to interact with substrates other than histones, including α-tubulin [75,76]. The inhibition of HDAC6 has been reported to ameliorate the severity of inherited neuropathy in animal models—such as Charcot–Marie–Tooth type 2—indicating, such as mentioned above, a possible role of acetylation and HDAC6 in the onset of neuropathies [76]. Nevertheless, much still needs to be elucidated about the role of post-transcriptional modifications on MT function and dynamics.
Two other essential bricks constitute the complex CTK structure: The microfilaments of actin and the intermediate filaments or neurofilaments. All of them combined, form a stable and functional network of polymers that are tightly associated and work in synergy.
Actin monomers polymerize forming a double helical structure, thinner (about 7nm) and more flexible than the MTs. In the periphery of the neuronal body, attached to the inner plasma membrane through anchoring proteins (ERM proteins), actin filaments form a meshwork together with actin-binding proteins and myosin motors. This so-called actomyosin cortex has an important role in protecting the cell shape against mechanical stress and in cell shape control [64]. In axons, however, actin ring-like structures bind to the inner membrane of the axon and are periodically arranged (~180 to 190 nanometers) along their shaft. In this way, MTs and actin jointly create a strong structure, able to resist the mechanical deformation forces exerted, in particular, on those axons that need to cover long distances (from hundreds of micrometer up to 1 m, depending on the cell type and on the species). Moreover, periodic actin ring organization has a very important role in stabilizing MT remodeling in growing and mature axons. In dendrites, instead, long actin filaments are positioned along the shaft [77,78]. The actin cytoskeleton is also responsible for the maintenance of dendritic spines shape, as well as for their dynamic morphological changes. The crosstalk of actin with dendritic MTs, together with the presence of some associated proteins (drebrin, end-binding EB3 or cortactin-binding protein 2), regulates the shape dynamics in spines during spinogenesis and throughout learning and memory processes [64,79]. During development, at the distal end of the incipient processes, complex networks of actin and a high number of actin-associated proteins work together with MTs to construct the polarized mature neuron. This complex machinery helps the exploratory filopodia and lamellipodia to detect the surrounding attractive or repellent cues present in the environment that will determine, in the end, the direction and the speed of the growing neurite until the final formation of a new synapse [80,81]. Therefore, MTs and actin must interact and be physically and functionally tightly coupled for the correct steering of axon and dendrite growth, spine plasticity and synapse formation [82].
An additional vital component of the cellular cytoskeleton is the intermediate filament. There are six major classes expressed in different cell types, all with a similar basic structure [83]. In neurons, neurofilaments (NF) constitute the class IV of intermediate filaments and are extremely long-lived proteins. NFs are 10 nm diameter twisted protein strands, whose composition (the relative subunit expression levels) varies at each neuron type at a specific stage of development. In fact, NFs are a combination of four different subunits: Light (NF-L), medium (NF-M) and heavy (NF-H) polypeptides, and the proteins α-internexin (present in the central nervous system) or peripherin (present only in the peripheral nervous system, or PNS). All three-polypeptide subunits differ highly on their aminoacidic composition at their head domain (amino-terminal end) and in particular, at the tail domain (carboxy-terminal end), which is also very variable in length. In addition, the presence of more or less post-translational modifications (mainly phosphorylation and glycosylation) contribute to the heterogeneity of these subunits [84,85]. NF structure is formed of the parallel side by side association of polypeptide dimers that in turn, arrange into staggered antiparallel tetramers. This particular organization of the NF tetramers produces a construct void of polarity, in contrast with the polar structures of actin filaments and MTs. The lateral association of eight tetramers creates cylindrical structures called unit length filaments, which join end-to-end to generate the final NF structure [83,86].
Their flexible fibrous structure allows the neuron to maintain its markedly asymmetric shape [87]. Neurofilaments are the most abundant CTK component in large myelinated caliber axons (with respect to perikarya and dendrites), where they have a role in developing and maintaining the three-dimensional array of the axoplasm. Moreover, neurofilaments are required for axon radial growth [88,89]. The carboxyl terminal of the filaments is subject to different post-translational modifications and protrude radially from the main filamentous structure. In particular, the side arms of the NF-M and NF-H subunits are responsible for keeping distant the neighboring filaments, in a degree that determines the diameter of the axon and, consequently, its final conduction velocity properties [83,90,91].
As already mentioned, the interaction of the three components of the cytoskeleton is fundamental for neuronal structural organization. Consequently, the proteins that allow the crosstalk among them are crucial, and present a possible direct or indirect target for neurotoxic drugs or neurological disease.
Spectraplakins are a family of large intracellular proteins with the peculiarity of being able to associate to all three filamentous components of the CTK [92,93]. In particular, dystonin (also known as bullous pemphigoid antigen 1, or BPAG1), is a very large spectraplakin protein present in different tissues such as the nervous system, skin and muscle. In the PNS, it is expressed primarily in sensory neurons, such as those of the DRG. Dystonin is essential for maintaining cytoskeletal organization and stability, organelle integrity, and intracellular transport. This protein is associated with the cytoskeleton, forming a bridge between F-actin and the intermediate filaments [94]. Loss of dystonin function causes dystonia musculorum (dt) in mice, a progressive loss of limb coordination that is the consequence of severe CTK disorganization in sensory neurons of the DRG [95]. Specifically, in DRG, there are several known subunits: Dystonin a1, a2 and a3. They present different N-terminal domains and different cellular distributions. For example, Dyst-a1 (also known as BPAG1n4) is present in axons, where it seems to be involved in retrograde transport mediated by dynactin [96,97]. Dyst-a2 possesses a transmembrane domain and has been found anchored to the external nuclear membrane [98], where it plays a role in perikaryon CTK organization, endoplasmic reticulum (ER) structure organization and endoplasmic reticulum-Golgi apparatus (ER-Golgi) vesicular transport [99]. Dyst-a3, however, localizes close to the plasma membrane [100].
4. Microtubules and CIPN
Considering the high compartmentalization of neurons and the central role of microtubules in cargo trafficking, most studies about neurotoxicity have focused on this class of cytoskeleton polymer [101,102,103,104,105,106]. Moreover, several neurodegenerative diseases of the peripheral nervous system present MT injury [107,108]. Microtubule function and/or structural perturbation also includes those affecting MAPs or other proteins able to interact with tubulin monomer or polymer.
4.1. Axonal Transport
As mentioned above, axonal transport allows the maintenance and restock of nerve endings with proteins, lipids and organelles (especially mitochondria). It also removes misfolded or damaged proteins that should be recycled [109,110].
Among all hypothesis proposed in the literature, impairment of axonal transport is one of the most plausible mechanisms that could lead to MTA-induced CIPN. Even if it is still unclear how these drugs alter axonal transport, it is known that they do not interfere by steric obstruction. For example, the binding site of taxanes and epothilones is localized on the inner surface of the MT, while motor proteins dock and move along the outer surface [111]. It is likely that the binding of both drugs induces a conformational change in the MT that alters the affinity between tubulin and motor proteins or other non-motor MAPs [112,113].
La Pointe et al. have evaluated the different effect of four main different classes of MTA (vincristine, eribulin, paclitaxel and ixabepilone) on axonal transport using vesicle the transport/squid axoplasm assay and the MT gliding assay [17]. They have hypothesized that the severity of neuropathy induced by these drugs is directly correlated with their ability to impair axonal transport. They have demonstrated that vincristine and ixabepilone reduce both anterograde and retrograde fast axonal transport, affecting also kinesin-1 velocity, while paclitaxel and eribulin impair only anterograde transport in a less significant manner and without affecting kinesin-1 movement.
The differences in neurotoxicity observed between paclitaxel and ixabepilone could be explained by their binding affinity. They are both MT-stabilizing agents, but ixabepilone binds to tubulin stronger than paclitaxel [114,115]. Moreover, their binding site to tubulin is not identical and for this reason, they could induce a different alteration in conformation and tubulin post-translational modification, especially in the c-terminal, a site that is important for motor proteins and MAP interaction and regulation [111,116,117].
Vincristine and eribulin are both MT destabilizing agents, but they impair axonal transport with different intensities. This difference could be also in this case explained by their different binding site: Eribulin binds only the plus-end of MT and exerts its destabilizing effect only here [118]. Conversely, vincristine binds along the entire MT and its effect destabilizes all the structure, altering the conformation of the MT and the interaction with motor proteins and MAPs [14,113].
4.2. Microtubule Polarity
As mentioned above, MT orientation is mixed in the cellular body and in dendrites, while it is highly polarized in axons. Here, MTs have the minus-end facing the cellular body and the plus-end facing the synapses. Alteration in this organization could lead to impaired axonal transport and consequently, axon degeneration and a decrease in conduction velocity [102,119,120]. In fact, Shemesh et al. have demonstrated that paclitaxel induces the reconfiguration of the MT from a highly polar oriented MT to a chaotic distribution [121]. This could be one of the possible causes of fast axonal transport impairment. How paclitaxel induces this alteration is still unclear, but two main hypotheses have been formulated. (1) Paclitaxel induces conformational changes that curve the MT in a loop. Until now, however, this alteration has been only observed in tubulin polymerization assays, but not in cell cultures or in vivo animal models [121,122,123]; (2) paclitaxel causes the formation of new nucleation sites that sequester tubulin dimers, from which newly assembled MTs extend in various directions [124]. Several research groups have also demonstrated the importance of MT conformation. In particular, a GTP/GDP (guanosine triphosphate/guanosine diphosphate) ratio alteration could induce conformational changes in the curvature of the MTs. In fact, the binding affinity of several MAPs and motor proteins is different on the base of MT curvature. For example, Nakata et al. have demonstrated that KIF5 (a kinesin-1) preferentially binds to GTP-rich regions of MT [125].
4.3. Microtubule-Associated Proteins
Microtubule-associated proteins (MAPs) are a class of proteins that interact with tubulin and allow MT to carry out all their functions. Paclitaxel impairment of axonal transport could be due to alteration of MAPs, or regulatory protein (for example kinases) expression or post-translational modifications [126,127,128]. Kinesin and dynein post-translational modifications have been reported in several inherited neurodegenerative diseases and this could be a valid mechanism in MTA-induced CIPN. One of the most studied modifications of kinesin is phosphorylation by JNK (c-Jun N-terminal kinase). This phosphorylation reduces the activity of kinesin. Moreover, the activation of JNK has been reported in non-cancer cells and in cortical neuron cultures after paclitaxel treatment [126,129]. This could explain the reduced axonal transport observed in paclitaxel-induced neurotoxicity.
Several other protein kinases regulate axonal transport through direct phosphorylation of motor proteins, adapter and cargos. Moreover, the dysregulation of axonal transport by kinases has been implicated in the pathogenesis of several neuropathies [130,131,132]. Both kinesin and dynein are regulated by protein kinases such as GSK3β, ERK1/2, JNK and Akt [127]. GSK3β activation induces the phosphorylation of kinesin and dynein motor proteins, reducing their movement along the MT. Interestingly, Gao et al. have demonstrated that paclitaxel increased activation of GSK3β in the spinal dorsal horn of rats and that paclitaxel-induced neuropathy is prevented when animals receive lithium treatment (a GSK3β inhibitor) [133].
As mentioned above, Tau is localized—particularly in the axons of mature neurons—and has been widely investigated due to its key role in neuronal development and in many neurodegenerative diseases [134,135,136]. Tau has two binding sites on the MT and one of these sites partially overlaps with the binding site of paclitaxel. Tau and Paclitaxel both suppress dynamic instability and increase stabilization of the MT, but Choi et al. have also demonstrated that they modulate the diameter of the MT and change the number of protofilaments by different amounts [137]. It is possible that, due to the overlapping binding site, paclitaxel competes with Tau for docking to the MT and induces a different conformational change in MT curvature and diameter. Samsonov et al. have shown that treatment with Paclitaxel reduced the ratio of Tau bound to the MT, confirming the competition to the same binding site [138]. Moreover, Park et al. have reported that polymorphisms in Tau-associated genes (i.e., GSK3β) may contribute to the development of paclitaxel-induced neuropathy [139]. They found a GSK3β polymorphism that increases Tau phosphorylation, reducing the rate of Tau association with the MT. Consequently, the MTs are less stable and more sensitive to paclitaxel. This could explain the role of tau phosphorylation in axonal damage and neuropathy induced by paclitaxel.
MAP1B is an MT-associated protein with important roles in development (axonal guidance and elongation) and function of the nervous system. In mature neurons, it participates in the regulation of the structure and physiology of dendritic spines. It binds to two subunits of tubulin and is involved in the nucleation and stabilization of the MT. It is reported that mutations or a knockout of the MAP1B gene change EB1 and EB3 interaction with the MT, leading to altered dynamics, directionality and curvature of the MT [140]. Brazil et al. have optimized a model of paclitaxel-induced peripheral neuropathy using Drosophila larvae and demonstrated that nociceptive sensitivity was associated with the disrupted organization of MAP1B/Futsch and an aberrant stabilization of peripheral sensory dendrites of class IV dendritic arborization nociceptors [141].
4.4. End-Tracking Proteins
Plus-end tracking proteins (+TIPs) are fundamental for the promotion and the stabilization of the growing MT [142]. +TIPs also have a central role in the interactions that occur between microtubules and actin [143,144,145].
Eribulin binds directly to the plus-end of the MT, its binding site is probably partially overlapping with some +TIP sites, and this could interfere with MT stability. O’Rurke et al. have demonstrated that the treatment with eribulin induces a dose-dependent reduction in the binding of the +TIP EB1 to the MT. The consequence was a reduction of MT stability and increased depolymerization [118].
Taxanes and vinca alkaloids also modulate the dynamics of MT plus-ends and disrupt EB protein localization. However, very little is known about the MTA action on +TIPs. Tubulin and microtubule conformational changes induced by taxanes and vinca alkaloids may alter +TIP interactions with tubulin or decrease the available EB binding sites at the microtubule plus-ends. Alternatively, MTAs could regulate EB-microtubule interaction indirectly via specific changes on EB protein phosphorylation [146].
Recently, Rovini et al. have demonstrated that paclitaxel treatment induces the loss of EB3-comet tails in cultured Aplysia neurons, with a subsequent reduction in MT dynamics and impaired axonal transport [146]. They have also reported that olesoxime partially reverts this alteration, preventing the delocalization of EB1 and EB3 proteins and preserving the MT dynamics.
4.5. Post-Translational Modification
In recent work, Van Helleputte et al. have demonstrated that the inhibition of histone deacetylase HDAC6 increases the acetylation of tubulin in several tissues of rodents and reduces the severity of the neuropathy induced by vincristine [147]. Increased tubulin acetylation induced by HDAC6 inhibitors partially restores MT stability and axonal transport. Moreover, tubulin acetylation increased the interaction and docking of motor proteins [76,147].
Similarly, Krukowski et al. demonstrated the same neuroprotective effect in an animal model treated with cisplatin [148]. The inhibition of HDAC6 increased α-tubulin acetylation in peripheral nerves, restoring axonal transport and mitochondrial function, and reverting mechanical allodynia induced by cisplatin.
4.6. Other Possible Microtubule-Correlated Pathways
Heat shock protein 27 (Hsp27) is involved in the reduction of reactive oxygen species during oxidative stress, anti-apoptotic activity under condition of chemical stress, and protein folding, but also interacts with actin and the MT [119]. The interaction with the MT is still poorly understood, but Hsp27 participates in the stabilization of the MT [149,150]. The role of Hsp27 in neurodegeneration has been reported in some inherited diseases. For example, mutations of HSP27 genes, inducing its higher affinity binding to the MT and consequently MT stabilization, are reported to cause Charcot–Marie–Tooth neuropathy [151]. A research group demonstrated that the overexpression of Hsp27 protects from paclitaxel-induced neuropathy. They proposed a different hypothesis, where a possible Hsp27 direct effect on the MT exists [119].
Mitofusins (Mfn1 and Mfn2) are proteins localized in the outer mitochondrial membrane and are involved in the fusion process of mitochondria. They are also involved in mitochondria transport along axons. Indeed, Mfn1 and Mfn2 interact with Miro1/Miro2 and the Milton proteins to form a complex that link mitochondria to kinesin motor proteins [152,153,154]. Mutations in Mfn2 cause Charcot–Marie–Tooth (CMT) type 2A, a disease characterized by degeneration of peripheral axons [155,156]. Bobylev et al. have demonstrated that cisplatin induces a reduction in the expression levels of Mfn2 [157]. Since cisplatin-induced peripheral neuropathy is linked to mitochondrial damage and alterations in their transport, it is possible that a reduction in Mfn2 expression could lead to impairment of mitochondria axonal transport. Yamashita et al. have also hypothesized that paclitaxel may induce peripheral neuropathy, due to changes in Mfn2 expression [158]. They have demonstrated that rats treated with paclitaxel show a reduction in Mfn2 expression levels in the spinal cord. This reduction is observed before the appearance of mechanical allodynia, suggesting that a reduction in Mfn2 expression contributes to paclitaxel-induced mechanical allodynia.
NF-κB is a transcriptional regulator that is physiologically inhibited by inhibitor kB alpha (IkBα). The activation of NF-κB occurs when the proteasome degrades IkBα and NF-κB translocates into the nucleus. In cancer cells, Bortezomib (BTZ) inhibits proteasome degradation of IkBα and, consequently NF-κB activation [159]. However, in neurons the situation is different. Alé et al. demonstrated that BTZ treatment induces the activation and nuclear translocation of NF-κB and consequently the increase of pro-inflammatory cytokines synthesis and cellular activation [22,160,161]. Pharmacological inhibition of NF-κB in neuron cultures reduces the degeneration of axons induced by BTZ and transgenic mice overexpressing IkBa suffer a less severe BTZ-induced neuropathy (BIPN). These data demonstrate a key role of NF-κB in BIPN, but the mechanism of NF-κB activation in neurons is still unclear. Several research groups have reported a correlation between the alteration in MT polymerization or axon damage and NF-κB activation, with a consequent increase in pro-inflammatory cytokines and inflammation [162,163,164].
5. Intermediate Filaments and CIPN
Because of the central role of intermediate filaments in maintaining cell integrity, axonal growth and caliber, and plasticity of the cytoskeleton, and considering their involvement in neurodegenerative disorders [165], these components of the cytoskeleton have been investigated in several papers regarding neurotoxicity. In the literature, there are data regarding expression, post-translational modifications and altered distributions of intermediate filaments. Meyer et al. have investigated, in three different papers, the effect of vincristine [166], oxaliplatin [167] and paclitaxel [168] on NF-H expression, studying also the protective effect of neurosteroids. Vincristine induced disorganization and a 44% decrease of NF-H immunostaining in rat sciatic nerves [166]. Interestingly, neurosteroids are able to reverse (to normal values) the vincristine-induced, decreased levels of NF-H, together with nerve conduction velocity, pain transmission abnormalities and intraepidermal nerve fiber density. It is important to underline that treatment of naïve rats with neurosteroids does not increase NF-H levels, suggesting that the effect of neurosteroids on NF-H may be indirect and that neurosteroids could affect the expression or activation levels of intracellular factors altered by vincristine.
On the other hand, Meyer et al. have highlighted that oxaliplatin induced a decrease of NF-H immunoreactivity in a rat sciatic nerve (59%) or the lumbar DRG (48%), together with sciatic nerve conduction velocity and peak amplitude reduction [167]. All these effects were reversed by allopregnanolone. This strict correlation between NF-H expression and electrophysiological parameters is in agreement with data obtained in each NF knock-out mouse [169]. In particular, Kriz et al., have demonstrated that low expression of NF-H induces axonal diameter reduction and consequently nerve conduction velocity slows down. In addition, Jamieson et al. have investigated the effect of different antineoplastic drugs on phosphorylated neurofilaments heavy chain (pNF-H) in the L5 dorsal root ganglia of adult female Wistar rats [170]. Oxaliplatin treatment induces a reduction of pNF-H immunoreactivity in neuronal cell bodies but it does not affect NF-H phosphorylation or expression in nerve fibers. Moreover, it is noteworthy that this endpoint is tightly correlated with the neurotoxicity of oxaliplatin, cisplatin and carboplatin. Conversely, it is not related to paclitaxel-induced neurotoxicity, suggesting that the reduction of pNF-H could be caused by an inhibition of NF-H kinase gene transcription (or induction of NF-H phosphatase), which is determined in a specific way by platinum-based drugs.
Paclitaxel (1 mg/kg for 7 days treatment), like vincristine and oxaliplatin, reduces (33%) NF-H immunostaining in treated rat sciatic nerve axons [168]. Paclitaxel-induced neuropathy and NF-H expression reduction is prevented by the natural neurosteroid 3α-androstanediol. Similarly to the effect of neurosteroids on vincristine-induced NF-H expression modulation, 3α-androstanediol also seems to modify the activation of transcription factors evoked by paclitaxel treatment. No paclitaxel-induced alterations of pNF-H expression in DRG neurons have been shown by Jamieson et al. [170].
Moreover, Alè et al. have demonstrated an alteration of neurofilament organization induced by bortezomib, both in in vitro and in vivo models [171]. In particular, in DRG neurons treated with 4 nM bortezomib, sensory neurons showed disruption of NF-H staining along neurites, whereas soma are characterized by their accumulation. DRG ganglia explanted from bortezomib-treated (2 mg/kg) mice show no alteration of NF-H mRNA expression, but an increase of the non-phosphorylated form of NF-H.
It is noteworthy that phosphorylation state of NFs is fundamental because this post-translational modification can perturb their own axonal transport, and consequently their distribution in neurons. In fact, many studies demonstrated that the rate of NF axonal transport is inversely correlated to their phosphorylation state [172]. In particular, C-terminal NF phosphorylation decreases the affinity for kinesin (anterograde transport) and increases their affinity for dynein (retrograde transport), whereas NF hypophosphorylation improves their anterograde dynein-mediated transport, as cargo of those microtubules are being translocated along axons [173]. At the same time though, NF phosphorylation could improve the NF-NF interaction, possibly making an NF macrostructure too large to be transported in retrograde. Moreover, the proper aggregation and distribution of neurofilaments can be modified by alteration of molecular motors or malfunction of microtubule dynamic structure, as already mentioned.
6. Microfilaments and CIPN
Surprisingly, a very small number of studies dealing with CIPN consider the effect of chemotherapeutics on microfilaments, despite their central role in maintaining the morphology of neurons and the numerous proteins with which they interact both in actin rings, actin waves, actin trails and hotspots [174].
Considering the little data present in the literature, the effect of antineoplastic drugs on microfilaments, and in particular, on actin rings and actin waves, should be deeply studied, considering the wide actin-microtubule crosstalk [64]. In fact, microfilaments or actin-binding proteins may be a target of antineoplastic drugs, which ultimately affect the rate of axonal microtubule bundling. Burnette et al. have indeed demonstrated that the actin ring in the initial axon segment, interacting with myosin, can align and promote the formation of the microtubule bundle [175]. Moreover, crosstalk between microfilaments and microtubules appears increasingly important in determining axon specification and outgrowth and in maintaining axonal mechanical protection.
James et al. have demonstrated in vitro, that paclitaxel and Cisplatin (CDDP) induce neurite degeneration, which affects the arrangement of the actin cytoskeleton in several neurons models, including DRG sensory neurons [176]. All the antineoplastic drugs tested reduced neurite outgrowth and branch number but, in particular, cisplatin neurotoxicity is mitigated by the reduction of RhoA activity. RhoA belongs to the Rho family small GTPases, which are regulatory molecules that link surface receptors to actin, as well as to microtubule cytoskeleton, and thus play a fundamental role in regulating neuronal morphology, dendritic arborization, spine morphogenesis and growth cone development [177]. It was demonstrated that: Rho inhibition improves axonal outgrowth and regeneration after injury [178] and; the Rho GTPases are involved in some neurodegenerative diseases [179].
Karademir et al., investigating the possible causes of different CIPN—induced by the two proteasome inhibitors BTZ and Carfilzomib (CFZ)—using a proteomic approach in a neural stem cell model, have demonstrated that among the most affected proteins (including microtubule) are actin and the actin-binding proteins [180]. In particular, BTZ and CFZ induce an actin density decrease, directly dependent on their neurotoxicity. Moreover, BTZ and CFZ are able to induce a transitory 2-fold increase of actin-related protein 2 (Arp2) expression, a protein that controls actin patch precursors in the PNS [181]. The increase of Arp2 expression after proteasome inhibitor treatment could be interpreted as a cellular response vs the destabilization of actin filaments. Moreover, interestingly BTZ but not CFZ increases some heat shock protein levels, including Hsp70, which interacts with actin and is involved in its transports to the proteasome [182].
In addition, in primary sensory neurons purified from Hsp27 transgenic mice, the overexpression of Hsp27 was proven to be effective in protecting from paclitaxel-induced neurite outgrowth inhibition [119]. Hsp27 is known to be an actin capping protein, able to bind to the terminal part of actin filaments, regulating their polymerization in several cell types. The same neuroprotection has been demonstrated in an in vivo experiment. Data suggest that the interaction between Hsp27 and the F-actin tips in the DRG could accelerate axonal growth and promote recovery from peripheral nerve damage. In this model, the Hsp27-induced axonal promoting effect could be correlated to inhibition of RhoA activity, as already demonstrated in rat cortical neurons [182].
7. Conclusions
Despite the development of new and more effective chemotherapies, most of the antineoplastic drugs used show a neurotoxic effect, that is dose-limiting and often determines discontinuation of treatment [1]. Furthermore, it is clear that the different classes of drugs, despite having a different antineoplastic effect, often induce CIPN characterized by similar and principally sensorial symptoms. At present, none of the strategies proposed, including complementary therapies with medicinal plants recently suggested [183], have proven effective in preventing or reducing nerve damage induced by antineoplastic drugs [1,27]. Starting from these concepts, this review has been focused on the changes induced by different chemotherapies on the neuronal cytoskeleton, a dynamic and diversified backbone in each neuron district, which is fundamental for neuronal structure and influences (with all three its components) all neurons functions. It is relevant to underline, that most of the papers regarding CIPN pathways are focused, independently of the chemotherapy drug under study, on the effect of antineoplastic agents on axon transport mediated by microtubules (which can perturb the organization of the other components of the cytoskeleton). From the data collected in this review, it is evident that there are some antineoplastic drug-induced effects that result independent of this mechanism. Tacchetti et al. studied the gene networks and functions of differentially expressed genes in the plasma cells of patients who developed bortezomib-thalidomide-dexamethasone-induced peripheral neuropathy, and demonstrated significant deregulation of processes involved in cytoskeleton rearrangement [184].
In order to study CIPN, cytoskeleton-related pathways that should be investigated are countless, considering the very high number of cytoskeleton binding proteins and of post-translational modifications of the MT, NF and microfilaments. In this context, taking into account the importance of crosstalk among the three components of the cytoskeleton, studies should focus on this aspect.
As mentioned in this review, several papers reported chemotherapy-induced modifications of the distribution of neurofilaments and of phosphorylation, in particular, of NF-H. However, in the literature, there is a lack of data regarding the activity of related kinases and phosphatases in CIPN models. The relevance of the C-terminal region of NF-H in mediating the interactions of NFs and other cytoskeleton components is well demonstrated [185,186,187]. Considering the fundamental role of neurofilaments in maintaining the stability of the axon (in particular, in determining the axon caliber in large fibers), in the future, it would be desirable to understand the effect of different classes of antineoplastic drugs on the mechanisms through which NF-H phosphorylation occurs, and the resulting effects on the other cytoskeleton components.
Based on the data in the literature regarding MT post-transcriptional modification, we think that alteration of MT lysine-40 acetylation is worthy of further study. In fact, it is a highly conserved modification, particularly present in the sensory neurons in mice, where it seems to modulate mechanosensation, among other functions. It is noteworthy to remark that MT acetylation has been reported relevant in a series of neuronal diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Rett syndrome and Charcot–Marie–Tooth (CMT) disease [188], while only some papers report a link between MT acetylation and CIPN. Moreover, in this context, only an indirect correlation of HDAC6 activity and CIPN has been studied. However, there are very few in vivo reports investigating the possible interaction between chemotherapy drugs and TAT1 activity. This interaction has been investigated only in vitro by Kalebic et al. [189].
The effect of antineoplastic drugs on microfilaments, still scarcely investigated, could be a very interesting goal. Despite the report that the actin ring, along the axon, stabilizes microtubule bundles, the pathway by which this phenomenon occurred is still not elucidated. Future CIPN studies should be focused on this aspect. In particular, spectraplakins could be an interesting target for neuroprotective studies, considering their ability to bind to all three components of the cytoskeleton. The importance of the study of the effect of chemotherapeutic agents on actin is even more evident, considering that in vitro, different classes of antineoplastic drug induce not only the reduction of neurite outgrowth but also the increase of neurite arborization (author’s personal observation), an event strictly modulated by actin filaments in growing neurites [190].
Finally, in the future, it would be desirable to develop pharmacological strategies based on a more in-depth knowledge of the effects that the different chemotherapeutic agents have on the ability of the three components of the cytoskeleton to crosstalk among themselves.
Author Contributions
A.M wrote “Microtubules and CIPN” and prepared Figure 1; C.M. wrote “Chemotherapy-induced peripheral neuropathy (CIPN)”, “CIPN models” and prepared Table 1; V.R.-M. wrote “Neuronal cytoskeleton” and contributed to the writing of “Conclusions”; G.N. wrote “Abstract”, “Intermediate filaments”, “Microfilaments and CIPN” and “Conclusions” and supervised the manuscript. All authors approved the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hershman, D.L.; Lacchetti, C.; Dworkin, R.H.; Lavoie Smith, E.M.; Bleeker, J.; Cavaletti, G.; Chauhan, C.; Gavin, P.; Lavino, A.; Lustberg, M.B.; et al. Prevention and Management of Chemotherapy-Induced Peripheral Neuropathy in Survivors of Adult Cancers: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2014, 32, 1941–1967. [Google Scholar] [CrossRef]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorg. Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef]
- Poruchynsky, M.S.; Komlodi-Pasztor, E.; Trostel, S.; Wilkerson, J.; Regairaz, M.; Pommier, Y.; Zhang, X.; Kumar Maity, T.; Robey, R.; Burotto, M.; et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 1571–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derry, W.B.; Wilson, L.; Jordan, M.A. Low potency of taxol at microtubule minus ends: Implications for its antimitotic and therapeutic mechanism. Cancer Res. 1998, 58, 1177–1184. [Google Scholar]
- Foley, E.A.; Kapoor, T.M. Microtubule attachment and spindle assembly checkpoint signalling at the kinetochore. Nat. Rev. Mol. Cell Biol. 2013, 14, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Vitale, I.; Buchmann, B.; Galluzzi, L.; Schwede, W.; Senovilla, L.; Skuballa, W.; Vivet, S.; Lichtner, R.B.; Vicencio, J.M.; et al. Improved Cellular Pharmacokinetics and Pharmacodynamics Underlie the Wide Anticancer Activity of Sagopilone. Cancer Res. 2008, 68, 5301–5308. [Google Scholar] [CrossRef] [Green Version]
- Addington, J.; Freimer, M. Chemotherapy-induced peripheral neuropathy: An update on the current understanding. F1000Research 2016, 5, 1466. [Google Scholar] [CrossRef]
- Starobova, H.; Vetter, I. Pathophysiology of Chemotherapy-Induced Peripheral Neuropathy. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyette-Davis, J.A.; Walters, E.T.; Dougherty, P.M. Mechanisms involved in the development of chemotherapy-induced neuropathy. Pain Manag. 2015, 5, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Miltenburg, N.C.; Boogerd, W. Chemotherapy-induced neuropathy: A comprehensive survey. Cancer Treat. Rev. 2014, 40, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Waseem, M.; Kaushik, P.; Tabassum, H.; Parvez, S. Role of Mitochondrial Mechanism in Chemotherapy-Induced Peripheral Neuropathy. Curr. Drug Metab. 2018, 19, 47–54. [Google Scholar] [CrossRef]
- Fukuda, Y.; Li, Y.; Segal, R.A. A Mechanistic Understanding of Axon Degeneration in Chemotherapy-Induced Peripheral Neuropathy. Front. Neurosci. 2017, 11, 481. [Google Scholar] [CrossRef]
- Liu, Y.-M.; Chen, H.-L.; Lee, H.-Y.; Liou, J.-P. Tubulin inhibitors: A patent review. Expert Opin. Ther. Pat. 2014, 24, 69–88. [Google Scholar] [CrossRef] [PubMed]
- Lobert, S.; Vulevic, B.; Correia, J.J. Interaction of Vinca Alkaloids with Tubulin: A Comparison of Vinblastine, Vincristine, and Vinorelbine. Biochemistry 1996, 35, 6806–6814. [Google Scholar] [CrossRef]
- Staff, N.P.; Grisold, A.; Grisold, W.; Windebank, A.J. Chemotherapy-induced peripheral neuropathy: A current review. Ann. Neurol. 2017, 81, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Benbow, S.J.; Cook, B.M.; Reifert, J.; Wozniak, K.M.; Slusher, B.S.; Littlefield, B.A.; Wilson, L.; Jordan, M.A.; Feinstein, S.C. Effects of Paclitaxel and Eribulin in Mouse Sciatic Nerve: A Microtubule-Based Rationale for the Differential Induction of Chemotherapy-Induced Peripheral Neuropathy. Neurotox. Res. 2016, 29, 299–313. [Google Scholar] [CrossRef]
- LaPointe, N.E.; Morfini, G.; Brady, S.T.; Feinstein, S.C.; Wilson, L.; Jordan, M.A. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: Implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology 2013, 37, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojima, I.; Chakravarty, S.; Inoue, T.; Lin, S.; He, L.; Horwitz, S.B.; Kuduk, S.D.; Danishefsky, S.J. A common pharmacophore for cytotoxic natural products that stabilize microtubules. Proc. Natl. Acad. Sci. USA 1999, 96, 4256–4261. [Google Scholar] [CrossRef] [Green Version]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Kerckhove, N.; Collin, A.; Condé, S.; Chaleteix, C.; Pezet, D.; Balayssac, D. Long-Term Effects, Pathophysiological Mechanisms, and Risk Factors of Chemotherapy-Induced Peripheral Neuropathies: A Comprehensive Literature Review. Front. Pharmacol. 2017, 8, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alé, A.; Bruna, J.; Navarro, X.; Udina, E. Neurotoxicity induced by antineoplastic proteasome inhibitors. Neurotoxicology 2014, 43, 28–35. [Google Scholar] [CrossRef]
- Curran, M.P.; McKeage, K. Bortezomib: A review of its use in patients with multiple myeloma. Drugs 2009, 69, 859–888. [Google Scholar] [CrossRef] [PubMed]
- Giannini, F.; Volpi, N.; Rossi, S.; Passero, S.; Fimiani, M.; Cerase, A. Thalidomide-induced neuropathy: A ganglionopathy? Neurology 2003, 60, 877–878. [Google Scholar] [CrossRef] [PubMed]
- Boyette-Davis, J.A.; Hou, S.; Abdi, S.; Dougherty, P.M. An updated understanding of the mechanisms involved in chemotherapy-induced neuropathy. Pain Manag. 2018, 8, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Bruna, J.; Marmiroli, P.; Cavaletti, G. Chemotherapy-induced peripheral neurotoxicity (CIPN): An update. Crit. Rev. Oncol. Hematol. 2012, 82, 51–77. [Google Scholar] [CrossRef] [PubMed]
- Sisignano, M.; Baron, R.; Scholich, K.; Geisslinger, G. Mechanism-based treatment for chemotherapy-induced peripheral neuropathic pain. Nat. Rev. Neurol. 2014, 10, 694–707. [Google Scholar] [CrossRef]
- Marmiroli, P.; Riva, B.; Pozzi, E.; Ballarini, E.; Lim, D.; Chiorazzi, A.; Meregalli, C.; Distasi, C.; Renn, C.L.; Semperboni, S.; et al. Susceptibility of different mouse strains to oxaliplatin peripheral neurotoxicity: Phenotypic and genotypic insights. PLoS ONE 2017, 12, e0186250. [Google Scholar] [CrossRef]
- Meregalli, C.; Chiorazzi, A.; Carozzi, V.A.; Canta, A.; Sala, B.; Colombo, M.; Oggioni, N.; Ceresa, C.; Foudah, D.; La Russa, F.; et al. Evaluation of tubulin polymerization and chronic inhibition of proteasome as citotoxicity mechanisms in bortezomib-induced peripheral neuropathy. Cell Cycle 2014, 13, 612–621. [Google Scholar] [CrossRef]
- Vichaya, E.G.; Chiu, G.S.; Krukowski, K.; Lacourt, T.E.; Kavelaars, A.; Dantzer, R.; Heijnen, C.J.; Walker, A.K. Mechanisms of chemotherapy-induced behavioral toxicities. Front. Neurosci. 2015, 9, 1–17. [Google Scholar] [CrossRef]
- Authier, N.; Fialip, J.; Eschalier, A.; Coudoré, F. Assessment of allodynia and hyperalgesia after cisplatin administration to rats. Neurosci. Lett. 2000, 291, 73–76. [Google Scholar] [CrossRef]
- Canta, A.; Chiorazzi, A.; Carozzi, V.A.; Meregalli, C.; Oggioni, N.; Bossi, M.; Rodriguez-Menendez, V.; Avezza, F.; Crippa, L.; Lombardi, R.; et al. Age-related changes in the function and structure of the peripheral sensory pathway in mice. Neurobiol. Aging 2016, 45, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Boehmerle, W.; Huehnchen, P.; Peruzzaro, S.; Balkaya, M.; Endres, M. Electrophysiological, behavioral and histological characterization of paclitaxel, cisplatin, vincristine and bortezomib-induced neuropathy in C57Bl/6 mice. Sci. Rep. 2015, 4, 6370. [Google Scholar] [CrossRef] [PubMed]
- Carozzi, V.A.; Canta, A.; Oggioni, N.; Sala, B.; Chiorazzi, A.; Meregalli, C.; Bossi, M.; Marmiroli, P.; Cavaletti, G. Neurophysiological and neuropathological characterization of new murine models of chemotherapy-induced chronic peripheral neuropathies. Exp. Neurol. 2010, 226, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Meregalli, C.; Ceresa, C.; Canta, A.; Carozzi, V.A.; Chiorazzi, A.; Sala, B.; Oggioni, N.; Lanza, M.; Letari, O.; Ferrari, F.; et al. CR4056, a new analgesic I2 ligand, is highly effective against bortezomib-induced painful neuropathy in rats. J. Pain Res. 2012, 5, 151. [Google Scholar] [CrossRef] [PubMed]
- Hoke, A.; Ray, M.; Höke, A.; Ray, M. Rodent Models of Chemotherapy-Induced Peripheral Neuropathy. ILAR J. 2014, 54, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Meregalli, C.; Fumagalli, G.; Alberti, P.; Canta, A.; Carozzi, V.A.; Chiorazzi, A.; Monza, L.; Pozzi, E.; Sandelius, Å.; Blennow, K.; et al. Neurofilament light chain as disease biomarker in a rodent model of chemotherapy induced peripheral neuropathy. Exp. Neurol. 2018, 307, 129–132. [Google Scholar] [CrossRef]
- Balayssac, D.; Ferrier, J.; Descoeur, J.; Ling, B.; Pezet, D.; Eschalier, A.; Authier, N. Chemotherapy-induced peripheral neuropathies: From clinical relevance to preclinical evidence. Expert Opin. Drug Saf. 2011, 10, 407–417. [Google Scholar] [CrossRef]
- Cavaletti, G.; Cavalletti, E.; Montaguti, P.; Oggioni, N.; De Negri, O.; Tredici, G. Effect on the peripheral nervous system of the short-term intravenous administration of paclitaxel in the rat. Neurotoxicology 1997, 18, 137–145. [Google Scholar]
- Nicolini, G.; Monfrini, M.; Scuteri, A. Axonal Transport Impairment in Chemotherapy-Induced Peripheral Neuropathy. Toxics 2015, 3, 322–341. [Google Scholar] [CrossRef] [Green Version]
- Scuteri, A.; Nicolini, G.; Miloso, M.; Bossi, M.; Cavaletti, G.; Windebank, A.J.; Tredici, G. Paclitaxel toxicity in post-mitotic dorsal root ganglion (DRG) cells. Anticancer Res. 2006, 26, 1065–1070. [Google Scholar]
- Grothe, C.; Unsicker, K. Neuron-enriched cultures of adult rat dorsal root ganglia: Establishment, characterization, survival, and neuropeptide expression in response to trophic factors. J. Neurosci. Res. 1987, 18, 539–550. [Google Scholar] [CrossRef]
- Geldof, A.A. Nerve-growth-factor-dependent neurite outgrowth assay; a research model for chemotherapy-induced neuropathy. J. Cancer Res. Clin. Oncol. 1995, 121, 657–660. [Google Scholar] [CrossRef]
- Windebank, A.J.; Blexrud, M.D. Characteristics of neurite outgrowth from rat spinal ganglia: Effects of serum and segmental level. J. Neuropathol. Exp. Neurol. 1986, 45, 683–691. [Google Scholar] [CrossRef]
- Meijering, E.; Jacob, M.; Sarria, J.-C.F.; Steiner, P.; Hirling, H.; Unser, M. Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry 2004, 58A, 167–176. [Google Scholar] [CrossRef]
- Henley, R.; Chandrasekaran, V.; Giulivi, C. Computing neurite outgrowth and arborization in superior cervical ganglion neurons. Brain Res. Bull. 2019, 144, 194–199. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Bradshaw, R.A. Activation of the Stat3 Signaling Pathway Is Required for Differentiation by Interleukin-6 in PC12-E2 Cells. J. Biol. Chem. 2000, 275, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Bilsland, J.; Rigby, M.; Young, L.; Harper, S. A rapid method for semi-quantitative analysis of neurite outgrowth from chick DRG explants using image analysis. J. Neurosci. Methods 1999, 92, 75–85. [Google Scholar] [CrossRef]
- Popova, D.; Jacobsson, S.O.P. A fluorescence microplate screen assay for the detection of neurite outgrowth and neurotoxicity using an antibody against βIII-tubulin. Toxicol. Vitr. 2014, 28, 411–418. [Google Scholar] [CrossRef]
- Konings, P.N.; Makkink, W.K.; van Delft, A.M.; Ruigt, G.S. Reversal by NGF of cytostatic drug-induced reduction of neurite outgrowth in rat dorsal root ganglia in vitro. Brain Res. 1994, 640, 195–204. [Google Scholar] [CrossRef]
- Malgrange, B.; Delrée, P.; Rigo, J.M.; Baron, H.; Moonen, G. Image analysis of neuritic regeneration by adult rat dorsal root ganglion neurons in culture: Quantification of the neurotoxicity of anticancer agents and of its prevention by nerve growth factor or basic fibroblast growth factor but not brain-derived neu. J. Neurosci. Methods 1994, 53, 111–122. [Google Scholar] [CrossRef]
- Livni, L.; Lees, J.G.; Barkl-Luke, M.E.; Goldstein, D.; Moalem-Taylor, G. Dorsal root ganglion explants derived from chemotherapy-treated mice have reduced neurite outgrowth in culture. Neurosci. Lett. 2019, 694, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Hamre, J.; Eldridge, S.; Behrsing, H.P.; Cutuli, F.M.; Mussio, J.; Davis, M. Multiparametric Image Analysis of Rat Dorsal Root Ganglion Cultures to Evaluate Peripheral Neuropathy-Inducing Chemotherapeutics. Toxicol. Sci. 2017, 156, kfw254. [Google Scholar] [CrossRef] [PubMed]
- Hoelting, L.; Klima, S.; Karreman, C.; Grinberg, M.; Meisig, J.; Henry, M.; Rotshteyn, T.; Rahnenführer, J.; Blüthgen, N.; Sachinidis, A.; et al. Stem Cell-Derived Immature Human Dorsal Root Ganglia Neurons to Identify Peripheral Neurotoxicants. Stem Cells Transl. Med. 2016, 5, 476–487. [Google Scholar] [CrossRef] [Green Version]
- Rana, P.; Luerman, G.; Hess, D.; Rubitski, E.; Adkins, K.; Somps, C. Utilization of iPSC-derived human neurons for high-throughput drug-induced peripheral neuropathy screening. Toxicol. Vitr. 2017, 45, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, H.E.; Wing, C.; Delaney, S.M.; Komatsu, M.; Dolan, M.E. Modeling Chemotherapeutic Neurotoxicity with Human Induced Pluripotent Stem Cell-Derived Neuronal Cells. PLoS ONE 2015, 10, e0118020. [Google Scholar] [CrossRef]
- Wing, C.; Komatsu, M.; Delaney, S.M.; Krause, M.; Wheeler, H.E.; Dolan, M.E. Application of stem cell derived neuronal cells to evaluate neurotoxic chemotherapy. Stem Cell Res. 2017, 22, 79–88. [Google Scholar] [CrossRef]
- Bhattacharya, M.R.C.; Gerdts, J.; Naylor, S.A.; Royse, E.X.; Ebstein, S.Y.; Sasaki, Y.; Milbrandt, J.; DiAntonio, A. A Model of Toxic Neuropathy in Drosophila Reveals a Role for MORN4 in Promoting Axonal Degeneration. J. Neurosci. 2012, 32, 5054–5061. [Google Scholar] [CrossRef] [Green Version]
- Lisse, T.S.; Middleton, L.J.; Pellegrini, A.D.; Martin, P.B.; Spaulding, E.L.; Lopes, O.; Brochu, E.A.; Carter, E.V.; Waldron, A.; Rieger, S. Paclitaxel-induced epithelial damage and ectopic MMP-13 expression promotes neurotoxicity in zebrafish. Proc. Natl. Acad. Sci. USA 2016, 113, E2189–E2198. [Google Scholar] [CrossRef] [Green Version]
- Podratz, J.L.; Staff, N.P.; Boesche, J.B.; Giorno, N.J.; Hainy, M.E.; Herring, S.A.; Klennert, M.T.; Milaster, C.; Nowakowski, S.E.; Krug, R.G., II; et al. An automated climbing apparatus to measure chemotherapy-induced neurotoxicity in Drosophila melanogaster. Fly 2013, 7, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Cairns, N.J.; Lee, V.M.Y.; Trojanowski, J.Q. The cytoskeleton in neurodegenerative diseases. J. Pathol. 2004, 204, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Hensel, N.; Claus, P. The Actin Cytoskeleton in SMA and ALS: How Does It Contribute to Motoneuron Degeneration? Neurosci. 2018, 24, 54–72. [Google Scholar] [CrossRef]
- Kung, A.L.; Zetterberg, A.; Sherwood, S.W.; Schimke, R.T. Cytotoxic effects of cell cycle phase specific agents: Result of cell cycle perturbation. Cancer Res. 1990, 50, 7307–7317. [Google Scholar] [CrossRef] [PubMed]
- Dogterom, M.; Koenderink, G.H. Actin–microtubule crosstalk in cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 38–54. [Google Scholar] [CrossRef]
- Kapitein, L.C.; Hoogenraad, C.C. Building the Neuronal Microtubule Cytoskeleton. Neuron 2015. [Google Scholar] [CrossRef] [PubMed]
- Conde, C.; Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef]
- Stepanova, T.; Slemmer, J.; Hoogenraad, C.C.; Lansbergen, G.; Dortland, B.; De Zeeuw, C.I.; Grosveld, F.; van Cappellen, G.; Akhmanova, A.; Galjart, N. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein). J. Neurosci. 2003, 23, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L.F. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Moughamian, A.J.; Osborn, G.E.; Lazarus, J.E.; Maday, S.; Holzbaur, E.L.F. Ordered Recruitment of Dynactin to the Microtubule Plus-End is Required for Efficient Initiation of Retrograde Axonal Transport. J. Neurosci. 2013, 33, 13190–13203. [Google Scholar] [CrossRef] [Green Version]
- Kapitein, L.C.; Yau, K.W.; Hoogenraad, C.C. Microtubule dynamics in dendritic spines. Methods Cell Biol. 2010, 97, 111–132. [Google Scholar] [CrossRef]
- Janke, C.; Kneussel, M. Tubulin post-translational modifications: Encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010, 33, 362–372. [Google Scholar] [CrossRef]
- Neumann, B.; Hilliard, M. Loss of MEC-17 leads to microtubule instability and axonal degeneration. Cell Rep. 2014. [Google Scholar] [CrossRef]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone Deacetylase 6 Inhibition Compensates for the Transport Deficit in Huntington’s Disease by Increasing Tubulin Acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef]
- Morley, S.J.; Qi, Y.; Iovino, L.; Andolfi, L.; Guo, D.; Kalebic, N.; Castaldi, L.; Tischer, C.; Portulano, C.; Bolasco, G.; et al. Acetylated tubulin is essential for touch sensation in mice. Elife 2016. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [Green Version]
- Van Helleputte, L.; Benoy, V.; Van Den Bosch, L. The role of histone deacetylase 6 (HDAC6) in neurodegeneration. Res. Rep. Biol. 2014, 5, 1. [Google Scholar] [CrossRef]
- Hahn, I.; Webb, S.E.D.; Pearce, S.P.; Prokop, A.; Qu, Y. Periodic actin structures in neuronal axons are required to maintain microtubules. Mol. Biol. Cell 2016. [Google Scholar] [CrossRef]
- Xu, K.; Zhong, G.; Zhuang, X. Actin, Spectrin, and Associated Proteins Form a Periodic Cytoskeletal Structure in Axons. Science (80-) 2013, 339, 452–456. [Google Scholar] [CrossRef]
- Nakahata, Y.; Yasuda, R. Plasticity of Spine Structure: Local Signaling, Translation and Cytoskeletal Reorganization. Front. Synaptic Neurosci. 2018, 10, 29. [Google Scholar] [CrossRef]
- Dent, E.W.; Gupton, S.L.; Gertler, F.B. The growth cone cytoskeleton in Axon outgrowth and guidance. Cold Spring Harb. Perspect. Biol. 2011. [Google Scholar] [CrossRef]
- Gomez, T.M.; Letourneau, P.C. Actin dynamics in growth cone motility and navigation. J. Neurochem. 2014, 129, 221–234. [Google Scholar] [CrossRef]
- Zhou, F.-Q.; Waterman-Storer, C.M.; Cohan, C.S. Focal loss of actin bundles causes microtubule redistribution and growth cone turning. J. Cell Biol. 2002, 157, 839–849. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef]
- Nakagawa, T.; Chen, J.; Zhang, Z.; Kanai, Y.; Hirokawa, N. Two distinct functions of the carboxyl-terminal tail domain of NF-M upon neurofilament assembly: Cross-bridge formation and longitudinal elongation of filaments. J. Cell Biol. 1995. [Google Scholar] [CrossRef]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments at a glance. J. Cell Sci. 2012. [Google Scholar] [CrossRef]
- Goldman, R.D.; Grin, B.; Mendez, M.G.; Kuczmarski, E.R. Intermediate filaments: Versatile building blocks of cell structure. Curr. Opin. Cell Biol. 2008, 20, 28–34. [Google Scholar] [CrossRef]
- Nixon, R.A.; Shea, T.B. Dynamics of neuronal intermediate filaments: A developmental perspective. Cell Motil. Cytoskeleton 1992, 22, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Eyer, J.; Peterson, A. Neurofilament-deficient axons and perikaryal aggregates in viable transgenic mice expressing a neurofilament-β-galactosidase fusion protein. Neuron 1994. [Google Scholar] [CrossRef]
- Hoffman, P.N.; Cleveland, D.W.; Griffin, J.W.; Landes, P.W.; Cowan, N.J.; Price, D.L. Neurofilament gene expression: A major determinant of axonal caliber. Proc. Natl. Acad. Sci. USA 1987, 84, 3472–3476. [Google Scholar] [CrossRef]
- Yin, X.; Crawford, T.O.; Griffin, J.W.; Tu, P.H.; Lee, V.M.; Li, C.; Roder, J.; Trapp, B.D. Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J. Neurosci. 1998, 18, 1953–1962. [Google Scholar] [CrossRef]
- Garcia, M.L.; Lobsiger, C.S.; Shah, S.B.; Deerinck, T.J.; Crum, J.; Young, D.; Ward, C.M.; Crawford, T.O.; Gotow, T.; Uchiyama, Y.; et al. NF-M is an essential target for the myelin-directed “outside-in” signaling cascade that mediates radial axonal growth. J. Cell Biol. 2003, 163, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Suozzi, K.C.; Wu, X.; Fuchs, E. Spectraplakins: Master orchestrators of cytoskeletal dynamics. J. Cell Biol. 2012, 197, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yue, J.; Wu, X. Spectraplakin family proteins—Cytoskeletal crosslinkers with versatile roles. J. Cell Sci. 2017. [Google Scholar] [CrossRef]
- Yang, Y.; Dowling, J.; Yu, Q.C.; Kouklis, P.; Cleveland, D.W.; Fuchs, E. An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell 1996. [Google Scholar] [CrossRef]
- Dalpé, G.; Leclerc, N.; Vallée, A.; Messer, A.; Mathieu, M.; De Repentigny, Y.; Kothary, R. Dystonin is essential for maintaining neuronal cytoskeleton organization. Mol. Cell. Neurosci. 1998. [Google Scholar] [CrossRef]
- Kapur, M.; Wang, W.; Maloney, M.T.; Millan, I.; Lundin, V.F.; Tran, T.A.; Yang, Y. Calcium tips the balance: A microtubule plus end to lattice binding switch operates in the carboxyl terminus of BPAG1n4. EMBO Rep. 2012. [Google Scholar] [CrossRef]
- Liu, J.J.; Ding, J.; Kowal, A.S.; Nardine, T.; Allen, E.; Delcroix, J.D.; Wu, C.; Mobley, W.; Fuchs, E.; Yang, Y. BPAG1n4 is essential for retrograde axonal transport in sensory neurons. J. Cell Biol. 2003. [Google Scholar] [CrossRef]
- Young, K.G.; Kothary, R. Dystonin/Bpag1 is a necessary endoplasmic reticulum/nuclear envelope protein in sensory neurons. Exp. Cell Res. 2008. [Google Scholar] [CrossRef]
- Ryan, S.D.; Ferrier, A.; Kothary, R. A novel role for the cytoskeletal linker protein dystonin in the maintenance of microtubule stability and the regulation of ER-Golgi transport. Bioarchitecture 2012. [Google Scholar] [CrossRef]
- Jefferson, J.J.; Leung, C.L.; Liem, R.K.H. Dissecting the sequence specific functions of alternative N-terminal isoforms of mouse bullous pemphigoid antigen 1. Exp. Cell Res. 2006. [Google Scholar] [CrossRef]
- Carlson, K.; Ocean, A.J. Peripheral Neuropathy with Microtubule-Targeting Agents: Occurrence and Management Approach. Clin. Breast Cancer 2011, 11, 73–81. [Google Scholar] [CrossRef]
- Cashman, C.R.; Höke, A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci. Lett. 2015, 596, 33–50. [Google Scholar] [CrossRef] [Green Version]
- Donovan, D. Management of Peripheral Neuropathy Caused by Microtubule Inhibitors. Clin. J. Oncol. Nurs. 2009, 13, 686–694. [Google Scholar] [CrossRef]
- Holzbaur, E.L.F.; Scherer, S.S. Microtubules, Axonal Transport, and Neuropathy. N. Engl. J. Med. 2011, 365, 2330–2332. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Swain, S.M. Peripheral Neuropathy Induced by Microtubule-Stabilizing Agents. J. Clin. Oncol. 2006, 24, 1633–1642. [Google Scholar] [CrossRef]
- Wozniak, K.M.; Vornov, J.J.; Wu, Y.; Nomoto, K.; Littlefield, B.A.; DesJardins, C.; Yu, Y.; Lai, G.; Reyderman, L.; Wong, N.; et al. Sustained Accumulation of Microtubule-Binding Chemotherapy Drugs in the Peripheral Nervous System: Correlations with Time Course and Neurotoxic Severity. Cancer Res. 2016, 76, 3332–3339. [Google Scholar] [CrossRef]
- Prior, R.; Van Helleputte, L.; Benoy, V.; Van Den Bosch, L. Defective axonal transport: A common pathological mechanism in inherited and acquired peripheral neuropathies. Neurobiol. Dis. 2017, 105, 300–320. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Millecamps, S.; Julien, J.-P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; LaPointe, N.; Bosco, D.A.; Brown, R.H.; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal Transport Defects in Neurodegenerative Diseases. J. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef]
- Khrapunovich-Baine, M.; Menon, V.; Yang, C.-P.H.; Northcote, P.T.; Miller, J.H.; Angeletti, R.H.; Fiser, A.; Horwitz, S.B.; Xiao, H. Hallmarks of Molecular Action of Microtubule Stabilizing Agents. J. Biol. Chem. 2011, 286, 11765–11778. [Google Scholar] [CrossRef] [Green Version]
- Elie-Caille, C.; Severin, F.; Helenius, J.; Howard, J.; Muller, D.J.; Hyman, A.A. Straight GDP-Tubulin Protofilaments Form in the Presence of Taxol. Curr. Biol. 2007, 17, 1765–1770. [Google Scholar] [CrossRef] [Green Version]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef]
- Kamath, K.; Jordan, M.A. Suppression of microtubule dynamics by epothilone B is associated with mitotic arrest. Cancer Res. 2003, 63, 6026–6031. [Google Scholar]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [Green Version]
- Khrapunovich-Baine, M.; Menon, V.; Verdier-Pinard, P.; Smith, A.B.; Angeletti, R.H.; Fiser, A.; Horwitz, S.B.; Xiao, H. Distinct Pose of Discodermolide in Taxol Binding Pocket Drives a Complementary Mode of Microtubule Stabilization. Biochemistry 2009, 48, 11664–11677. [Google Scholar] [CrossRef] [Green Version]
- Uchimura, S.; Oguchi, Y.; Hachikubo, Y.; Ishiwata, S.; Muto, E. Key residues on microtubule responsible for activation of kinesin ATPase. EMBO J. 2010, 29, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, B.; Yang, C.-P.H.; Sharp, D.; Horwitz, S.B. Eribulin disrupts EB1-microtubule plus-tip complex formation. Cell Cycle 2014, 13, 3218–3221. [Google Scholar] [CrossRef] [Green Version]
- Chine, V.B.; Au, N.P.B.; Kumar, G.; Ma, C.H.E. Targeting Axon Integrity to Prevent Chemotherapy-Induced Peripheral Neuropathy. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef]
- Topp, K.S.; Tanner, K.D.; Levine, J.D. Damage to the cytoskeleton of large diameter sensory neurons and myelinated axons in vincristine-induced painful peripheral neuropathy in the rat. J. Comp. Neurol. 2000, 424, 563–576. [Google Scholar] [CrossRef]
- Shemesh, O.A.; Spira, M.E. Paclitaxel induces axonal microtubules polar reconfiguration and impaired organelle transport: Implications for the pathogenesis of paclitaxel-induced polyneuropathy. Acta Neuropathol. 2010, 119, 235–248. [Google Scholar] [CrossRef]
- Hendricks, M.; Jesuthasan, S. PHR regulates growth cone pausing at intermediate targets through microtubule disassembly. J. Neurosci. 2009, 29, 6593–6598. [Google Scholar] [CrossRef]
- Letourneau, P.C.; Ressler, A.H. Inhibition of neurite initiation and growth by taxol. J. Cell Biol. 1984, 98, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Rohena, C.C.; Mooberry, S.L. Recent progress with microtubule stabilizers: New compounds, binding modes and cellular activities. Nat. Prod. Rep. 2014, 31, 335–355. [Google Scholar] [CrossRef]
- Nakata, T.; Yorifuji, H. Morphological evidence of the inhibitory effect of taxol on the fast axonal transport. Neurosci. Res. 1999, 35, 113–122. [Google Scholar] [CrossRef]
- DeBerg, H.A.; Blehm, B.H.; Sheung, J.; Thompson, A.R.; Bookwalter, C.S.; Torabi, S.F.; Schroer, T.A.; Berger, C.L.; Lu, Y.; Trybus, K.M.; et al. Motor Domain Phosphorylation Modulates Kinesin-1 Transport. J. Biol. Chem. 2013, 288, 32612–32621. [Google Scholar] [CrossRef]
- Gibbs, K.L.; Greensmith, L.; Schiavo, G. Regulation of Axonal Transport by Protein Kinases. Trends Biochem. Sci. 2015, 40, 597–610. [Google Scholar] [CrossRef] [Green Version]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule Acetylation Promotes Kinesin-1 Binding and Transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [Green Version]
- Morfini, G.; Pigino, G.; Szebenyi, G.; You, Y.; Pollema, S.; Brady, S.T. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat. Neurosci. 2006, 9, 907–916. [Google Scholar] [CrossRef]
- Chevalier-Larsen, E.; Holzbaur, E.L.F. Axonal transport and neurodegenerative disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2006, 1762, 1094–1108. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.-A.; Rizzo, V.; Puthanveettil, S. V Pathologies of axonal transport in neurodegenerative diseases. Transl. Neurosci. 2012, 3, 355–372. [Google Scholar] [CrossRef]
- Tourtellotte, W.G. Axon Transport and Neuropathy: Relevant Perspectives on the Etiopathogenesis of Familial Dysautonomia. Am. J. Pathol. 2016, 186, 489–499. [Google Scholar] [CrossRef]
- Gao, M.; Yan, X.; Weng, H.-R. Inhibition of glycogen synthase kinase 3beta activity with lithium prevents and attenuates paclitaxel-induced neuropathic pain. Neuroscience 2013, 254, 301–311. [Google Scholar] [CrossRef] [Green Version]
- Hutton, M.; Lewis, J.; Dickson, D.; Yen, S.H.; McGowan, E. Analysis of tauopathies with transgenic mice. Trends Mol. Med. 2001, 7, 467–470. [Google Scholar] [CrossRef]
- Iqbal, K.; Alonso, A.d.C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.-X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in Alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [Green Version]
- McKee, A.C.; Cantu, R.C.; Nowinski, C.J.; Hedley-Whyte, E.T.; Gavett, B.E.; Budson, A.E.; Santini, V.E.; Lee, H.-S.; Kubilus, C.A.; Stern, R.A. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 2009, 68, 709–735. [Google Scholar] [CrossRef]
- Choi, M.C.; Chung, P.J.; Song, C.; Miller, H.P.; Kiris, E.; Li, Y.; Wilson, L.; Feinstein, S.C.; Safinya, C.R. Paclitaxel suppresses Tau-mediated microtubule bundling in a concentration-dependent manner. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3456–3463. [Google Scholar] [CrossRef]
- Samsonov, A.; Yu, J.-Z.; Rasenick, M.; Popov, S. V Tau interaction with microtubules in vivo. J. Cell Sci. 2004, 117, 6129–6141. [Google Scholar] [CrossRef]
- Park, S.B.; Kwok, J.B.; Loy, C.T.; Friedlander, M.L.; Lin, C.S.Y.; Krishnan, A.V.; Lewis, C.R.; Kiernan, M.C. Paclitaxel-induced neuropathy: Potential association of MAPT and GSK3B genotypes. BMC Cancer 2014, 14, 993. [Google Scholar] [CrossRef]
- Tortosa, E.; Galjart, N.; Avila, J.; Sayas, C.L. MAP1B regulates microtubule dynamics by sequestering EB1/3 in the cytosol of developing neuronal cells. EMBO J. 2013, 32, 1293–1306. [Google Scholar] [CrossRef] [Green Version]
- Brazill, J.M.; Cruz, B.; Zhu, Y.; Zhai, R.G. Nmnat mitigates sensory dysfunction in a Drosophila model of paclitaxel-induced peripheral neuropathy. Dis. Model. Mech. 2018, 11, dmm032938. [Google Scholar] [CrossRef] [PubMed]
- Akhmanova, A.; Steinmetz, M.O. Tracking the ends: A dynamic protein network controls the fate of microtubule tips. Nat. Rev. Mol. Cell Biol. 2008, 9, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Akhmanova, A.; Steinmetz, M.O. Microtubule +TIPs at a glance. J. Cell Sci. 2010, 123, 3415–3419. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Chang, F. Shaping the actin cytoskeleton using microtubule tips. Curr. Opin. Cell Biol. 2007, 19, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Dugina, V.; Alieva, I.; Khromova, N.; Kireev, I.; Gunning, P.W.; Kopnin, P. Interaction of microtubules with the actin cytoskeleton via cross-talk of EB1-containing +TIPs and γ-actin in epithelial cells. Oncotarget 2016, 7, 72699–72715. [Google Scholar] [CrossRef] [Green Version]
- Rovini, A.; Carré, M.; Bordet, T.; Pruss, R.M.; Braguer, D. Olesoxime prevents microtubule-targeting drug neurotoxicity: Selective preservation of EB comets in differentiated neuronal cells. Biochem. Pharmacol. 2010, 80, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Van Helleputte, L.; Kater, M.; Cook, D.P.; Eykens, C.; Rossaert, E.; Haeck, W.; Jaspers, T.; Geens, N.; Vanden Berghe, P.; Gysemans, C.; et al. Inhibition of histone deacetylase 6 (HDAC6) protects against vincristine-induced peripheral neuropathies and inhibits tumor growth. Neurobiol. Dis. 2018, 111, 59–69. [Google Scholar] [CrossRef]
- Krukowski, K.; Ma, J.; Golonzhka, O.; Laumet, G.O.; Gutti, T.; van Duzer, J.H.; Mazitschek, R.; Jarpe, M.B.; Heijnen, C.J.; Kavelaars, A. HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain 2017, 158, 1126–1137. [Google Scholar] [CrossRef]
- Lavoie, J.N.; Lambert, H.; Hickey, E.; Weber, L.A.; Landry, J. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol. Cell. Biol. 1995, 15, 505–516. [Google Scholar] [CrossRef]
- Pichon, S.; Bryckaert, M.; Berrou, E. Control of actin dynamics by p38 MAP kinase-Hsp27 distribution in the lamellipodium of smooth muscle cells. J. Cell Sci. 2004, 117, 2569–2577. [Google Scholar] [CrossRef]
- Kalmar, B.; Innes, A.; Wanisch, K.; Kolaszynska, A.K.; Pandraud, A.; Kelly, G.; Abramov, A.Y.; Reilly, M.M.; Schiavo, G.; Greensmith, L. Mitochondrial deficits and abnormal mitochondrial retrograde axonal transport play a role in the pathogenesis of mutant Hsp27-induced Charcot Marie Tooth Disease. Hum. Mol. Genet. 2017, 26, 3313–3326. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two mitofusin proteins, mammalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef]
- Rojo, M.; Legros, F.; Chateau, D.; Lombès, A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 2002, 115, 1663–1674. [Google Scholar]
- Lawson, V.H.; Graham, B.V.; Flanigan, K.M. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology 2005, 65, 197–204. [Google Scholar] [CrossRef]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef] [Green Version]
- Bobylev, I.; Joshi, A.R.; Barham, M.; Neiss, W.F.; Lehmann, H.C. Depletion of Mitofusin-2 Causes Mitochondrial Damage in Cisplatin-Induced Neuropathy. Mol. Neurobiol. 2018, 55, 1227–1235. [Google Scholar] [CrossRef]
- Yamashita, Y.; Irie, K.; Kochi, A.; Kimura, N.; Hayashi, T.; Matsuo, K.; Myose, T.; Sano, K.; Nakano, T.; Takase, Y.; et al. Involvement of Charcot-Marie-Tooth disease gene mitofusin 2 expression in paclitaxel-induced mechanical allodynia in rats. Neurosci. Lett. 2017, 653, 337–340. [Google Scholar] [CrossRef]
- Ghosh, S.; Dass, J.F.P. Study of pathway cross-talk interactions with NF-κB leading to its activation via ubiquitination or phosphorylation: A brief review. Gene 2016, 584, 97–109. [Google Scholar] [CrossRef]
- Alé, A.; Bruna, J.; Calls, A.; Karamita, M.; Haralambous, S.; Probert, L.; Navarro, X.; Udina, E. Inhibition of the neuronal NFκB pathway attenuates bortezomib-induced neuropathy in a mouse model. Neurotoxicology 2016, 55, 58–64. [Google Scholar] [CrossRef]
- Hunot, S.; Brugg, B.; Ricard, D.; Michel, P.P.; Muriel, M.-P.; Ruberg, M.; Faucheux, B.A.; Agid, Y.; Hirsch, E.C. Nuclear translocation of NF-B is increased in dopaminergic neurons of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1997, 94, 7531–7536. [Google Scholar] [CrossRef]
- Jackman, R.W.; Rhoads, M.G.; Cornwell, E.; Kandarian, S.C. Microtubule-mediated NF-kappaB activation in the TNF-alpha signaling pathway. Exp. Cell Res. 2009, 315, 3242–3249. [Google Scholar] [CrossRef]
- Rai, A.; Kapoor, S.; Singh, S.; Chatterji, B.P.; Panda, D. Transcription factor NF-κB associates with microtubules and stimulates apoptosis in response to suppression of microtubule dynamics in MCF-7 cells. Biochem. Pharmacol. 2015, 93, 277–289. [Google Scholar] [CrossRef]
- Spencer, W.; Kwon, H.; Crépieux, P.; Leclerc, N.; Lin, R.; Hiscott, J. Taxol selectively blocks microtubule dependent NF-kappaB activation by phorbol ester via inhibition of IkappaBalpha phosphorylation and degradation. Oncogene 1999, 18, 495–505. [Google Scholar] [CrossRef]
- Lépinoux-Chambaud, C.; Eyer, J. Review on intermediate filaments of the nervous system and their pathological alterations. Histochem. Cell Biol. 2013, 140, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Meyer, L.; Patte-Mensah, C.; Taleb, O.; Mensah-Nyagan, A.G. Cellular and functional evidence for a protective action of neurosteroids against vincristine chemotherapy-induced painful neuropathy. Cell. Mol. Life Sci. 2010, 67, 3017–3034. [Google Scholar] [CrossRef]
- Meyer, L.; Patte-Mensah, C.; Taleb, O.; Mensah-Nyagan, A.G. Allopregnanolone prevents and suppresses oxaliplatin-evoked painful neuropathy: Multi-parametric assessment and direct evidence. Pain 2011, 152, 170–181. [Google Scholar] [CrossRef]
- Meyer, L.; Patte-Mensah, C.; Taleb, O.; Mensah-Nyagan, A.G. Neurosteroid 3α-Androstanediol Efficiently Counteracts Paclitaxel-Induced Peripheral Neuropathy and Painful Symptoms. PLoS ONE 2013, 8, e80915. [Google Scholar] [CrossRef]
- Kriz, J.; Zhu, Q.; Julien, J.P.; Padjen, A.L. Electrophysiological properties of axons in mice lacking neurofilament subunit genes: Disparity between conduction velocity and axon diameter in absence of NF-H. Brain Res. 2000, 885, 32–44. [Google Scholar] [CrossRef]
- Jamieson, S.M.F.; Subramaniam, J.; Liu, J.J.; Jong, N.N.; Ip, V.; Connor, B.; McKeage, M.J. Oxaliplatin-Induced Loss of Phosphorylated Heavy Neurofilament Subunit Neuronal Immunoreactivity in Rat Drg Tissue. Mol. Pain 2009, 5, 66. [Google Scholar] [CrossRef]
- Alé, A.; Bruna, J.; Herrando, M.; Navarro, X.; Udina, E. Toxic Effects of Bortezomib on Primary Sensory Neurons and Schwann Cells of Adult Mice. Neurotox. Res. 2015, 27, 430–440. [Google Scholar] [CrossRef]
- Perrot, R.; Berges, R.; Bocquet, A.; Eyer, J. Review of the Multiple Aspects of Neurofilament Functions, and their Possible Contribution to Neurodegeneration. Mol. Neurobiol. 2008, 38, 27–65. [Google Scholar] [CrossRef]
- Motil, J.; Chan, W.K.H.; Dubey, M.; Chaudhury, P.; Pimenta, A.; Chylinski, T.M.; Ortiz, D.T.; Shea, T.B. Dynein mediates retrograde neurofilament transport within axons and anterograde delivery of NFs from perikarya into axons: Regulation by multiple phosphorylation events. Cell Motil. Cytoskeleton 2006, 63, 266–286. [Google Scholar] [CrossRef]
- Dubey, P.; Jorgenson, K.; Roy, S. Actin Assemblies in the Axon Shaft—Some Open Questions. Curr. Opin. Neurobiol. 2018, 51, 163–167. [Google Scholar] [CrossRef]
- Burnette, D.T.; Ji, L.; Schaefer, A.W.; Medeiros, N.A.; Danuser, G.; Forscher, P. Myosin II Activity Facilitates Microtubule Bundling in the Neuronal Growth Cone Neck. Dev. Cell 2008, 15, 163–169. [Google Scholar] [CrossRef] [Green Version]
- James, S.E.; Burden, H.; Burgess, R.; Xie, Y.; Yang, T.; Massa, S.M.; Longo, F.M.; Lu, Q. Anti-cancer drug induced neurotoxicity and identification of Rho pathway signaling modulators as potential neuroprotectants. Neurotoxicology 2008, 29, 605–612. [Google Scholar] [CrossRef]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef]
- Madura, T.; Kubo, T.; Tanag, M.; Matsuda, K.; Tomita, K.; Yano, K.; Hosokawa, K. The Rho-Associated Kinase Inhibitor Fasudil Hydrochloride Enhances Neural Regeneration after Axotomy in the Peripheral Nervous System. Plast. Reconstr. Surg. 2007, 119, 526–535. [Google Scholar] [CrossRef]
- DeGeer, J.; Lamarche-Vane, N. Rho GTPases in neurodegeneration diseases. Exp. Cell Res. 2013, 319, 2384–2394. [Google Scholar] [CrossRef]
- Karademir, B.; Sari, G.; Jannuzzi, A.T.; Musunuri, S.; Wicher, G.; Grune, T.; Mi, J.; Hacioglu-Bay, H.; Forsberg-Nilsson, K.; Bergquist, J.; et al. Proteomic approach for understanding milder neurotoxicity of Carfilzomib against Bortezomib. Sci. Rep. 2018, 8, 16318. [Google Scholar] [CrossRef]
- Chou, F.-S.; Wang, P.-S. The Arp2/3 complex is essential at multiple stages of neural development. Neurogenes 2016, 3, e1261653. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.H.E.; Omura, T.; Cobos, E.J.; Latrémolière, A.; Ghasemlou, N.; Brenner, G.J.; van Veen, E.; Barrett, L.; Sawada, T.; Gao, F.; et al. Accelerating axonal growth promotes motor recovery after peripheral nerve injury in mice. J. Clin. Investig. 2011, 121, 4332–4347. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.-Y.; Liu, C.-T.; Su, Y.-L.; Chen, S.-Y.; Chen, Y.-H.; Tsai, M.-Y. A review of complementary therapies with medicinal plants for chemotherapy-induced peripheral neuropathy. Complement. Ther. Med. 2019, 42, 226–232. [Google Scholar] [CrossRef]
- Tacchetti, P.; Terragna, C.; Galli, M.; Zamagni, E.; Petrucci, M.T.; Pezzi, A.; Montefusco, V.; Martello, M.; Tosi, P.; Baldini, L.; et al. Bortezomib- and thalidomide-induced peripheral neuropathy in multiple myeloma: Clinical and molecular analyses of a phase 3 study. Am. J. Hematol. 2014, 89, 1085–1091. [Google Scholar] [CrossRef]
- Shea, T.B.; Beermann, M.L. Respective roles of neurofilaments, microtubules, MAP1B, and tau in neurite outgrowth and stabilization. Mol. Biol. Cell 1994, 5, 863–875. [Google Scholar] [CrossRef]
- Shea, T.B.; Chan, W.K.-H. Regulation of neurofilament dynamics by phosphorylation. Eur. J. Neurosci. 2008, 27, 1893–1901. [Google Scholar] [CrossRef]
- Shea, T.B.; Lee, S. Neurofilament phosphorylation regulates axonal transport by an indirect mechanism: A merging of opposing hypotheses. Cytoskeleton 2011, 68, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Kelley, T.; Rymut, S. Broader implications: Biological and clinical significance of microtubule acetylation. Cell Health Cytoskelet. 2015, 71. [Google Scholar] [CrossRef]
- Kalebic, N.; Martinez, C.; Perlas, E.; Hublitz, P.; Bilbao-Cortes, D.; Fiedorczuk, K.; Andolfo, A.; Heppenstall, P.A. Tubulin acetyltransferase αTAT1 destabilizes microtubules independently of its acetylation activity. Mol. Cell. Biol. 2013, 33, 1114–1123. [Google Scholar] [CrossRef]
- Leite, S.C.; Sampaio, P.; Sousa, V.F.; Nogueira-Rodrigues, J.; Pinto-Costa, R.; Peters, L.L.; Brites, P.; Sousa, M.M. The Actin-Binding Protein α-Adducin Is Required for Maintaining Axon Diameter. Cell Rep. 2016, 15, 490–498. [Google Scholar] [CrossRef]
Figure 1.
Neuronal cytoskeleton.


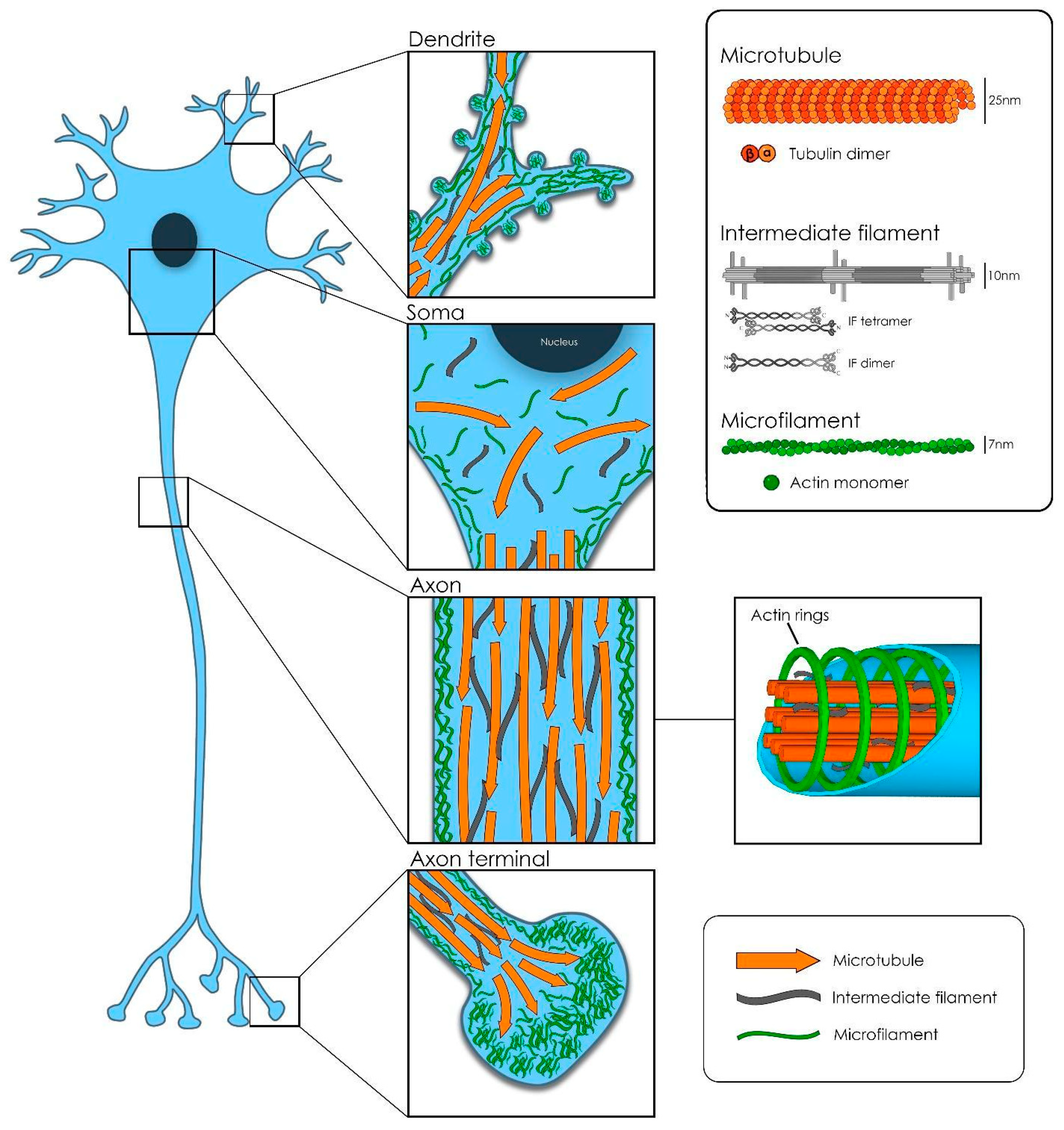{kind=link}
Table 1.
Summary of antineoplastic and neurotoxic mechanisms in chemotherapy-induced peripheral neurotoxicity. DRG: Dorsal root ganglia; NGF: Nerve growth factor; TRP: Transient receptor potential channels.
Table 1.
Summary of antineoplastic and neurotoxic mechanisms in chemotherapy-induced peripheral neurotoxicity. DRG: Dorsal root ganglia; NGF: Nerve growth factor; TRP: Transient receptor potential channels.
| Chemotherapy Agent | Antineoplastic Mechanisms | CIPN Pathophysiology |
|---|---|---|
| Taxanes | Microtubule damage that impairs mitotic spindle formation in cancer cells [4,5,6]. | Activation of caspases, oxidative stress on peripheral neuronal and non-neuronal cells, mitotoxicity, inhibition of anterograde fast axonal transport, prevention of microtubule disassembly, alteration of both activity and expression of voltage-gated ion channels in the DRG; immune activation in the DRG and peripheral nerves, and microglial activation in the spinal cord; TRP upregulation in DRG [7,8,9,10,11,12]. |
| Vinca alkaloids | Binding to free tubulin dimers close to the GTP-binding sites and induction of cell death by inhibition of microtubule assembly [13,14]. | Increase of microtubule depolymerization and inhibition of the hydrolysis of GTP; membrane excitability, inflammation, axonal transport impairment; mitochondria and glial function alterations; differential expression of voltage-gated ion channels, alteration of neurotransmission, impairment of axonal transport, increased production and release of proinflammatory cytokine and chemokines [8,9,11,12,15]. |
| Eribulin | Suppression of microtubule dynamic instability at low concentration and promotion of microtubule disassembly at high concentration [16]. | Reduction of anterograde fast axonal transport; reduction of kinesin-dependent transport in axon [17]. |
| Epothilones | Stabilization of microtubules, leading to apoptosis in cancer cells [18]. | Microtubule stabilization; reduction of kinesin-dependent transport [17]. |
| Platinum compounds | Platinum-DNA adduct formation, alterations in transmembrane receptors and channels that lead to cell cycle arrest and apoptosis [19,20]. | Accumulation of platinum atom in the DRG sensory neurons, nuclear and mitochondrial DNA damage, oxidative stress and channellopathy; TRP channels affected; axonal transport impairment; activation of caspases and protein kinase, glial cell activation, increased production and release of proinflammatory cytokine and chemokines [7,8,9,11,12,21]. |
| Proteasome inhibitors | Inhibition of proteasome activity, which results in protein aggregate accumulation in tumor cells, cell cycle arrest and apoptosis [22,23]. | Protein aggregate accumulation in soma neurons, alteration of physiological turnover of axonal proteins, axonal transport impairment; damage to neuronal mitochondria and organelles and Schwann cell microtubule stabilization, oxidative stress; activation of TRP channels; inflammation and glial cell activation; alterations of neurotransmission [9,10,11,12,15]. |
| Thalidomide | Immunomodulation and antiangiogenic effects, down-regulation of tumor necrosis factor alpha (TNFα) [24]. | Capillary damage due to its antiangiogenic activity in nerve fibers; downregulation of TNFα and inhibition of NF-κB that interferes with NGF activity [10,21]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Malacrida, A.; Meregalli, C.; Rodriguez-Menendez, V.; Nicolini, G. Chemotherapy-Induced Peripheral Neuropathy and Changes in Cytoskeleton. Int. J. Mol. Sci. 2019, 20, 2287. https://doi.org/10.3390/ijms20092287
AMA Style
Malacrida A, Meregalli C, Rodriguez-Menendez V, Nicolini G. Chemotherapy-Induced Peripheral Neuropathy and Changes in Cytoskeleton. International Journal of Molecular Sciences. 2019; 20(9):2287. https://doi.org/10.3390/ijms20092287
Chicago/Turabian StyleMalacrida, Alessio, Cristina Meregalli, Virginia Rodriguez-Menendez, and Gabriella Nicolini. 2019. "Chemotherapy-Induced Peripheral Neuropathy and Changes in Cytoskeleton" International Journal of Molecular Sciences 20, no. 9: 2287. https://doi.org/10.3390/ijms20092287
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.