Lipopolysaccharide-Induced Matrix Metalloproteinase-9 Expression Associated with Cell Migration in Rat Brain Astrocytes

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. LPS Induced MMP-9 Expression through Transcription and Translation
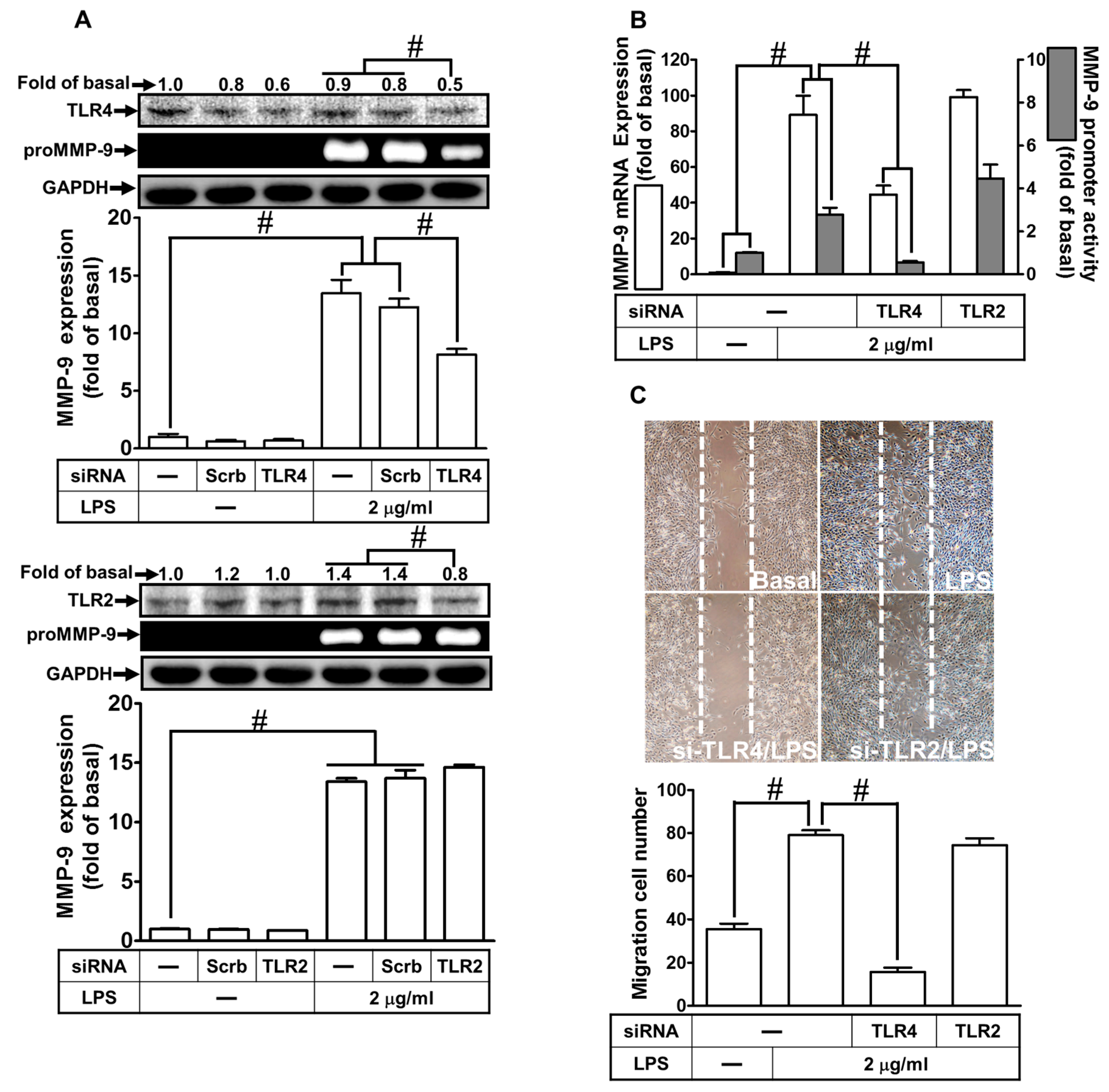
2.2. LPS Induced proMMP-9 Expression via TLR4
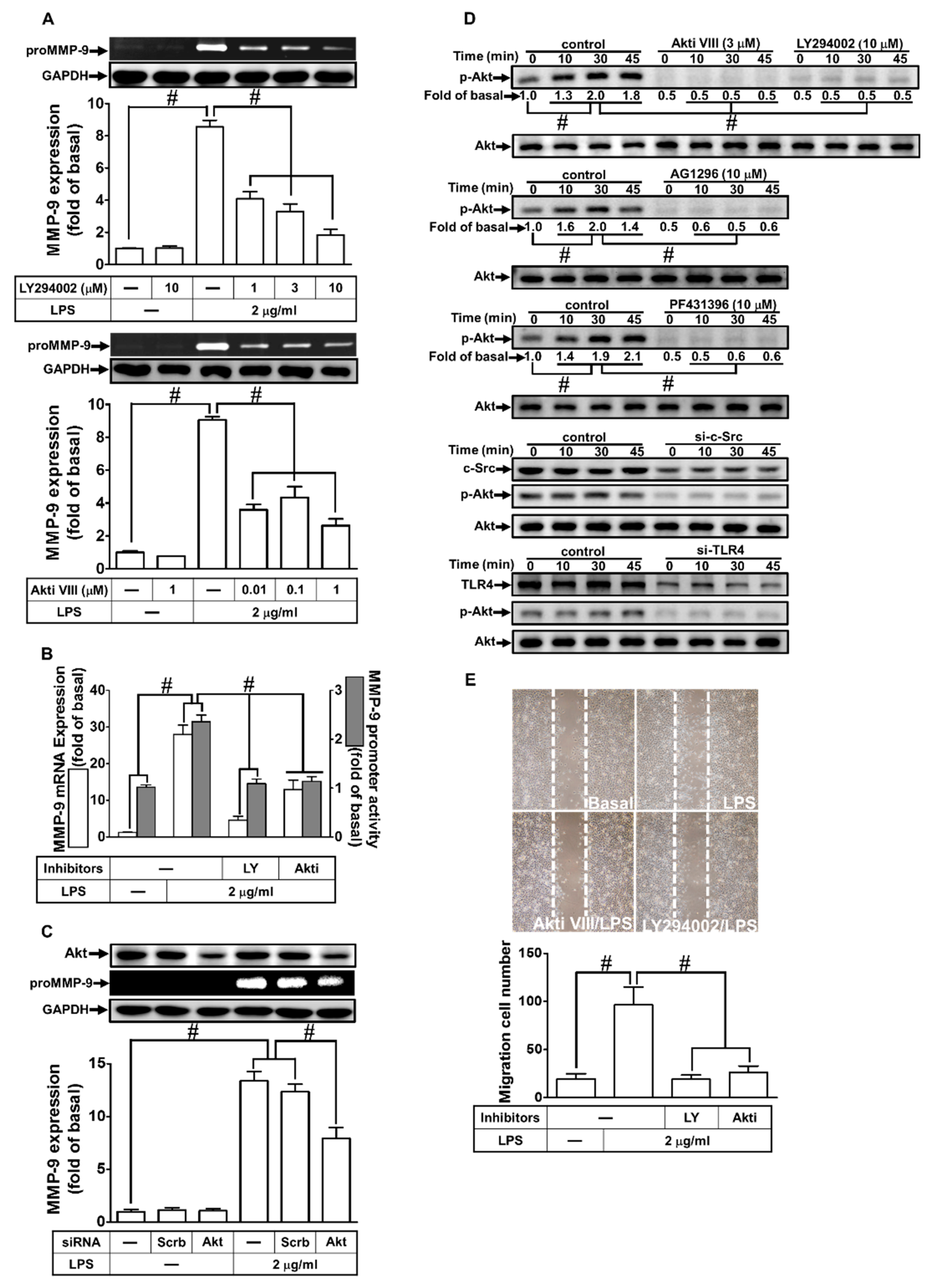
2.3. LPS-Induced proMMP-9 Expression Was Mediated via c-Src Activation
2.4. LPS Induced proMMP-9 Expression via Pyk2 Phosphorylation
2.5. LPS Induced proMMP-9 Expression through PDGFR Activation
2.6. PI3K/Akt Were Involved in LPS-Induced proMMP-9 Expression
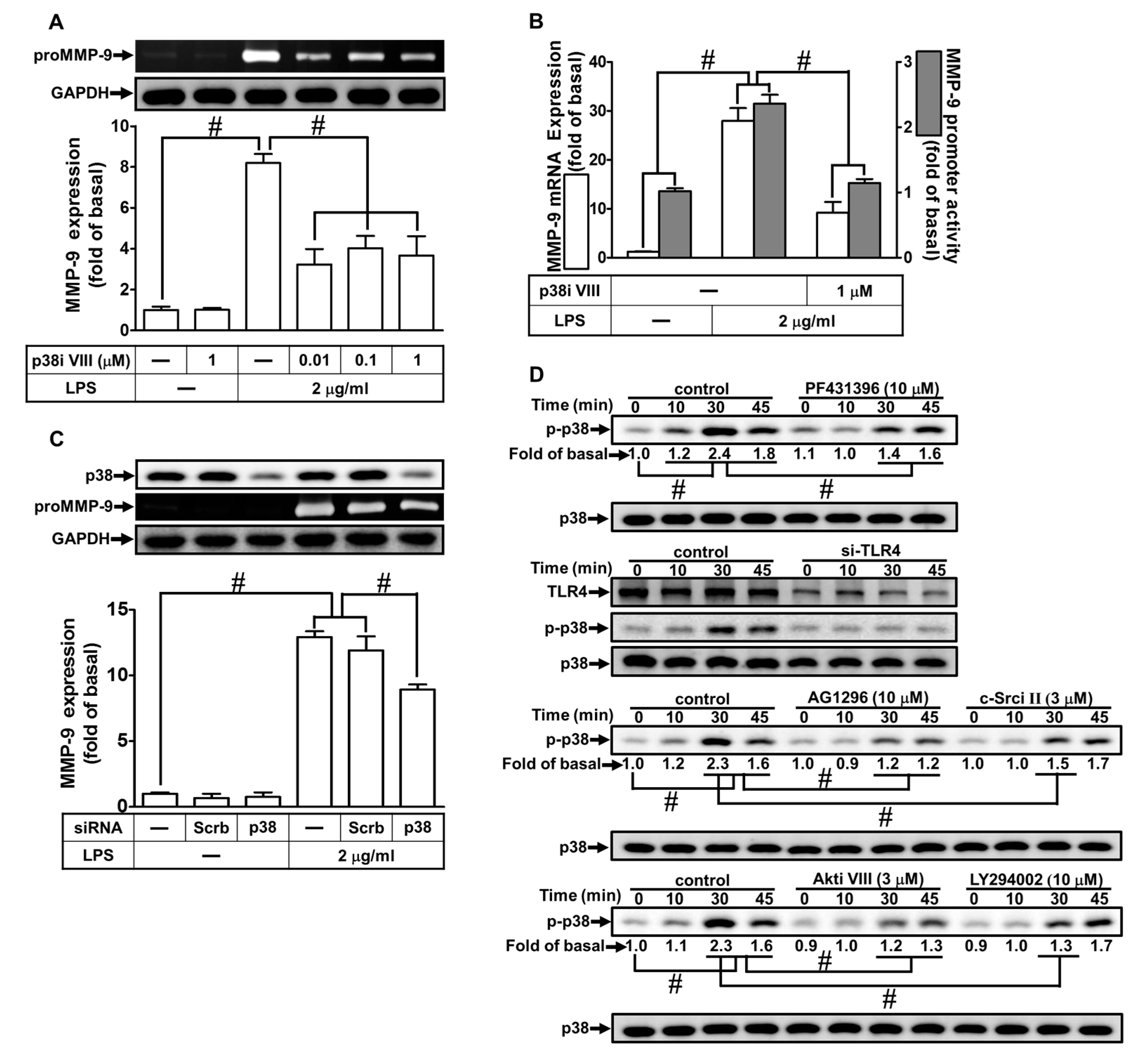
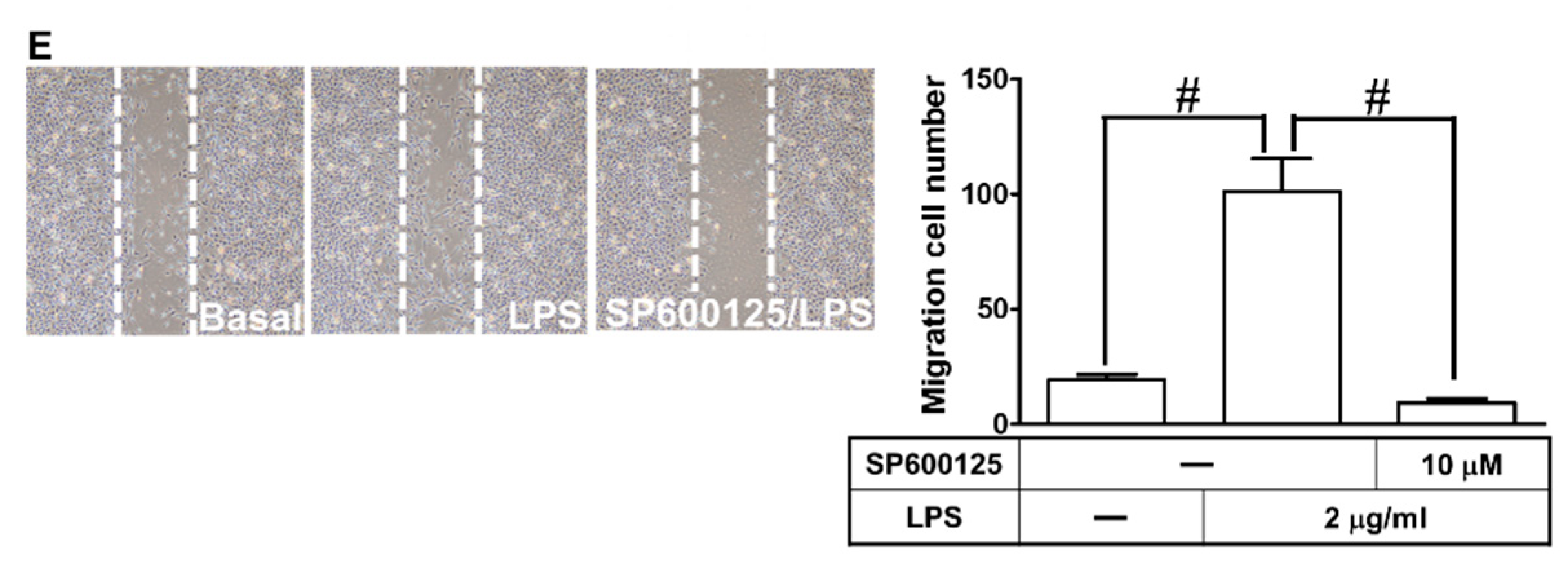
2.7. LPS Induced proMMP-9 Expression via MAPKs
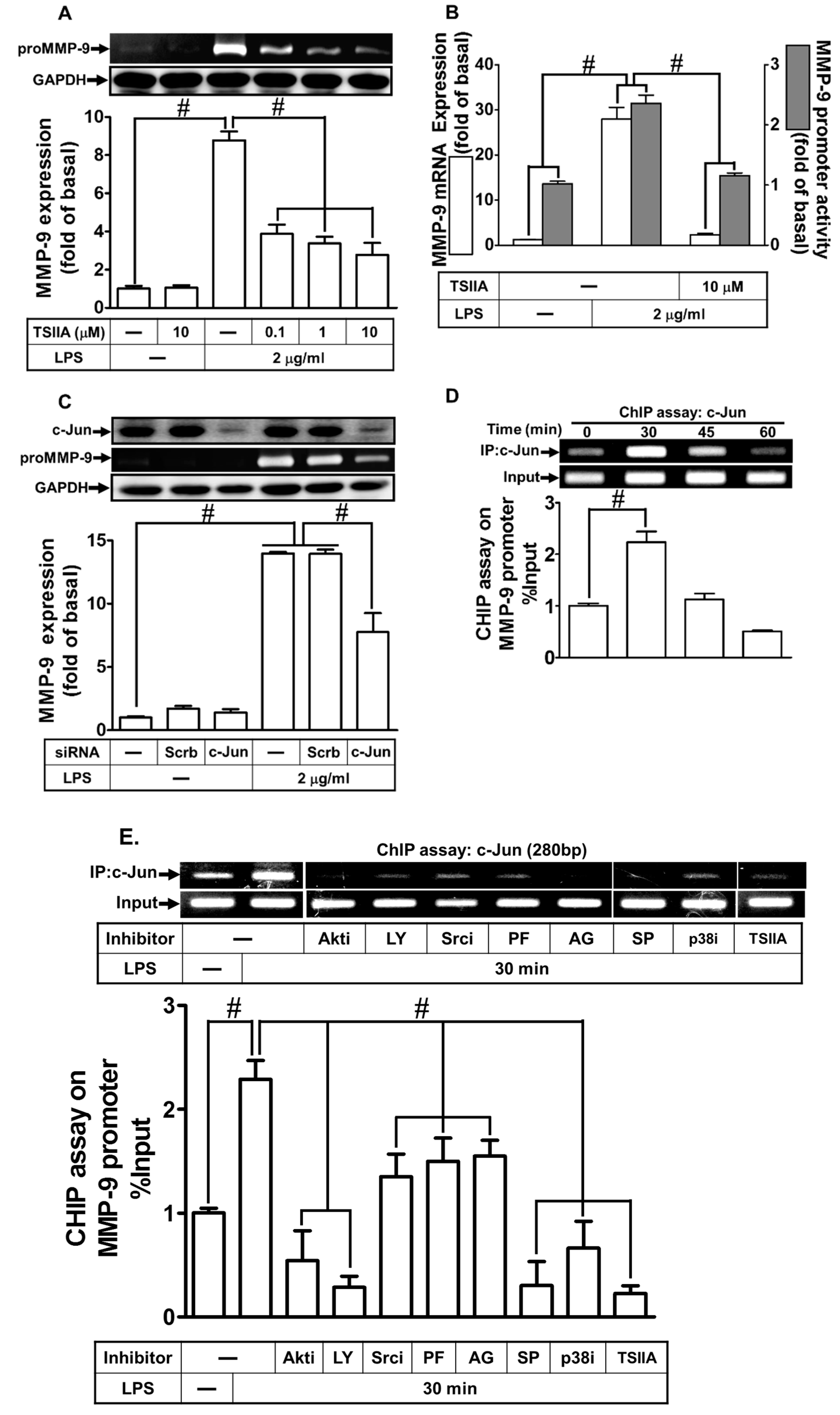
2.8. LPS Induced proMMP-9 Expression via AP-1
3. Discussion
4. Materials and Methods
4.1. Antibodies and Inhibitors
4.2. Cell Culture and Treatment
4.3. Protein Preparation and Western Blotting
4.4. MMP geLatin Zymography
4.5. Transient Transfection with siRNA
4.6. Total RNA Extraction and Real-Time PCR Analysis
- 5′-AGTTTGGTGTCGCGGAGCAC-3′ (sense);
- 5′-TACATGAGCGCTTCCGGCAC-3′ (antisense);
- 5′-CGCTCTGCATTTCTTCAAGGACGGT-3′-tetramethylrhodamine (TAMRA, Probe)
- 5′-(AACTTTGGCATCGTGGAAGG)-3′ (sense);
- 5′-(GTGGATGCAGGGATGATGTTC)-3′ (antisense);
- 5′- TGACCACAGTCCATGCCATCACTGC-3′-TAMRA (Probe).
4.7. Rat MMP-9 Promoter Reporter Gene Assay
4.8. Cell Migration Assay
4.9. Chromatin Immunoprecipitation (ChIP) Assay
4.10. Analysis of Data
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nag, S. Morphology and properties of astrocytes. Methods Mol. Biol. Clifton N. J. 2011, 686, 69–100. [Google Scholar] [CrossRef]
- Takemoto, T.; Ishihara, Y.; Ishida, A.; Yamazaki, T. Neuroprotection elicited by nerve growth factor and brain-derived neurotrophic factor released from astrocytes in response to methylmercury. Environ. Toxicol. Pharmacol. 2015, 40, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Li, J.R.; Chen, W.Y.; Ou, Y.C.; Lai, C.Y.; Hu, Y.H.; Wu, C.C.; Chang, C.J.; Chen, C.J. Disruption of in vitro endothelial barrier integrity by Japanese encephalitis virus-Infected astrocytes. Glia 2015, 63, 1915–1932. [Google Scholar] [CrossRef] [PubMed]
- Garwood, C.J.; Ratcliffe, L.E.; Simpson, J.E.; Heath, P.R.; Ince, P.G.; Wharton, S.B. Review: Astrocytes in Alzheimer’s disease and other age-associated dementias: A supporting player with a central role. Neuropathol. Appl. Neurobiol. 2017, 43, 281–298. [Google Scholar] [CrossRef]
- Ludwin, S.K.; Rao, V.; Moore, C.S.; Antel, J.P. Astrocytes in multiple sclerosis. Mult. Scler. 2016, 22, 1114–1124. [Google Scholar] [CrossRef]
- Wang, Q.; Jie, W.; Liu, J.H.; Yang, J.M.; Gao, T.M. An astroglial basis of major depressive disorder? An overview. Glia 2017, 65, 1227–1250. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Serotonin 1A Receptors on Astrocytes as a Potential Target for the Treatment of Parkinson’s Disease. Curr. Med. Chem. 2016, 23, 686–700. [Google Scholar] [CrossRef]
- Rosenberg, G.A. Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 2009, 8, 205–216. [Google Scholar] [CrossRef]
- Turner, R.J.; Sharp, F.R. Implications of MMP9 for Blood Brain Barrier Disruption and Hemorrhagic Transformation Following Ischemic Stroke. Front. Cell. Neurosci. 2016, 10, 56. [Google Scholar] [CrossRef] [Green Version]
- Kaminari, A.; Tsilibary, E.C.; Tzinia, A. A New Perspective in Utilizing MMP-9 as a Therapeutic Target for Alzheimer’s Disease and Type 2 Diabetes Mellitus. J. Alzheimers Dis. 2018, 64, 1–16. [Google Scholar] [CrossRef]
- Yang, C.M.; Lin, C.C.; Lee, I.T.; Lin, Y.H.; Yang, C.M.; Chen, W.J.; Jou, M.J.; Hsiao, L.D. Japanese encephalitis virus induces matrix metalloproteinase-9 expression via a ROS/c-Src/PDGFR/PI3K/Akt/MAPKs-dependent AP-1 pathway in rat brain astrocytes. J. Neuroinflamm. 2012, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppert, D.; Leib, S.L.; Grygar, C.; Miller, K.M.; Schaad, U.B.; Hollander, G.A. Matrix metalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during bacterial meningitis: Association with blood-brain barrier damage and neurological sequelae. Clin. Infect. Dis. 2000, 31, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Van den Steen, P.E.; Sang, Q.X.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Candelario-Jalil, E.; Yang, Y.; Rosenberg, G.A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 2009, 158, 983–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Han, Q. Efficacy of GM6001 as an adjuvant to ceftriaxone in a neonatal rat model of Streptococcus pneumoniae meningitis. Acta Neurobiol. Exp. Wars 2014, 74, 489–496. [Google Scholar] [PubMed]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Molteni, M.; Gemma, S.; Rossetti, C. The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediat. Inflamm. 2016, 2016, 6978936. [Google Scholar] [CrossRef] [Green Version]
- Cho, R.L.; Yang, C.C.; Lee, I.T.; Lin, C.C.; Chi, P.L.; Hsiao, L.D.; Yang, C.M. Lipopolysaccharide induces ICAM-1 expression via a c-Src/NADPH oxidase/ROS-dependent NF-kappaB pathway in human pulmonary alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L639–L657. [Google Scholar] [CrossRef] [Green Version]
- Anand, A.R.; Cucchiarini, M.; Terwilliger, E.F.; Ganju, R.K. The tyrosine kinase Pyk2 mediates lipopolysaccharide-induced IL-8 expression in human endothelial cells. J. Immunol. 2008, 180, 5636–5644. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.F.; Wang, H.; Fan, Y.; Shi, H.J.; Wang, Q.M.; Chen, B.R.; Khurwolah, M.R.; Long, Q.Q.; Wang, S.B.; Wang, Z.M.; et al. Epigallocatechin-3-Gallate Inhibits Matrix Metalloproteinase-9 and Monocyte Chemotactic Protein-1 Expression Through the 67-kappaDa Laminin Receptor and the TLR4/MAPK/NF-kappaB Signalling Pathway in Lipopolysaccharide-Induced Macrophages. Cell. Physiol. Biochem. 2017, 43, 926–936. [Google Scholar] [CrossRef]
- Slomiany, B.L.; Slomiany, A. Role of LPS-elicited signaling in triggering gastric mucosal inflammatory responses to H. pylori: Modulatory effect of ghrelin. Inflammopharmacology 2017, 25, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Hsiao, L.D.; Yang, C.M.; Lin, C.C. Thrombin Enhanced Matrix Metalloproteinase-9 Expression and Migration of SK-N-SH Cells via PAR-1, c-Src, PYK2, EGFR, Erk1/2 and AP-1. Mol. Neurobiol. 2017, 54, 3476–3491. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.C.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. Galangin Inhibits Thrombin-Induced MMP-9 Expression in SK-N-SH Cells via Protein Kinase-Dependent NF-kappaB Phosphorylation. Int. J. Mol. Sci. 2018, 19, 4084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.E.; Chang, M.Y.; Wei, J.Y.; Chen, Y.J.; Maa, M.C.; Leu, T.H. Berberine reduces Toll-like receptor-mediated macrophage migration by suppression of Src enhancement. Eur. J. Pharmacol. 2015, 757, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Lee, I.T.; Chi, P.L.; Hsieh, H.L.; Cheng, S.E.; Hsiao, L.D.; Liu, C.J.; Yang, C.M. C-Src/Jak2/PDGFR/PKCdelta-dependent MMP-9 induction is required for thrombin-stimulated rat brain astrocytes migration. Mol. Neurobiol. 2014, 49, 658–672. [Google Scholar] [CrossRef]
- Tsai, C.L.; Chen, W.C.; Lee, I.T.; Chi, P.L.; Cheng, S.E.; Yang, C.M. c-Src-dependent transactivation of PDGFR contributes to TNF-alpha-induced MMP-9 expression and functional impairment in osteoblasts. Bone 2014, 60, 186–197. [Google Scholar] [CrossRef]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Yang, C.C.; Lin, C.C.; Chien, P.T.; Hsiao, L.D.; Yang, C.M. Thrombin/Matrix Metalloproteinase-9-Dependent SK-N-SH Cell Migration is Mediated Through a PLC/PKC/MAPKs/NF-kappaB Cascade. Mol. Neurobiol. 2016, 53, 5833–5846. [Google Scholar] [CrossRef]
- Yang, C.M.; Lee, I.T.; Lin, C.C.; Wang, C.H.; Cherng, W.J.; Hsiao, L.D. c-Src-dependent MAPKs/AP-1 activation is involved in TNF-alpha-induced matrix metalloproteinase-9 expression in rat heart-derived H9c2 cells. Biochem. Pharmacol. 2013, 85, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Dutta, G.; Zhang, P.; Liu, B. The lipopolysaccharide Parkinson’s disease animal model: Mechanistic studies and drug discovery. Fundam. Clin. Pharmacol. 2008, 22, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Brandtzaeg, P.; Bjerre, A.; Ovstebo, R.; Brusletto, B.; Joo, G.B.; Kierulf, P. Neisseria meningitidis lipopolysaccharides in human pathology. J. Endotoxin Res. 2001, 7, 401–420. [Google Scholar] [CrossRef] [PubMed]
- Niranjan, R.; Nagarajan, R.; Hanif, K.; Nath, C.; Shukla, R. LPS induces mediators of neuroinflammation, cell proliferation, and GFAP expression in human astrocytoma cells U373MG: The anti-inflammatory and anti-proliferative effect of guggulipid. Neurol. Sci. 2014, 35, 409–414. [Google Scholar] [CrossRef]
- Tarassishin, L.; Suh, H.S.; Lee, S.C. LPS and IL-1 differentially activate mouse and human astrocytes: Role of CD14. Glia 2014, 62, 999–1013. [Google Scholar] [CrossRef] [Green Version]
- Vivekanantham, S.; Shah, S.; Dewji, R.; Dewji, A.; Khatri, C.; Ologunde, R. Neuroinflammation in Parkinson’s disease: Role in neurodegeneration and tissue repair. Int. J. Neurosci. 2015, 125, 717–725. [Google Scholar] [CrossRef]
- Xiang, W.; Chao, Z.Y.; Feng, D.Y. Role of Toll-like receptor/MYD88 signaling in neurodegenerative diseases. Rev. Neurosci. 2015, 26, 407–414. [Google Scholar] [CrossRef]
- Dikic, I.; Tokiwa, G.; Lev, S.; Courtneidge, S.A.; Schlessinger, J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature 1996, 383, 547–550. [Google Scholar] [CrossRef]
- Chien, P.T.; Lin, C.C.; Hsiao, L.D.; Yang, C.M. c-Src/Pyk2/EGFR/PI3K/Akt/CREB-activated pathway contributes to human cardiomyocyte hypertrophy: Role of COX-2 induction. Mol. Cell. Endocrinol. 2015, 409, 59–72. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Lennartsson, J. The PDGF/PDGFR pathway as a drug target. Mol. Asp. Med. 2018, 62, 75–88. [Google Scholar] [CrossRef]
- Perez, J.; Torres, R.A.; Rocic, P.; Cismowski, M.J.; Weber, D.S.; Darley-Usmar, V.M.; Lucchesi, P.A. PYK2 signaling is required for PDGF-dependent vascular smooth muscle cell proliferation. Am. J. Physiol. Cell Physiol. 2011, 301, C242–C251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.W.; Lin, C.C.; Lin, W.N.; Liang, K.C.; Luo, S.F.; Wu, C.B.; Wang, S.W.; Yang, C.M. TNF-alpha induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3K/Akt cascade and promotion of NF-kappaB/p300 binding in human tracheal smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L799–L812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.Y.; Kuo, C.T.; Lin, C.C.; Hsieh, H.L.; Yang, C.M. IL-1beta induces expression of matrix metalloproteinase-9 and cell migration via a c-Src-dependent, growth factor receptor transactivation in A549 cells. Br. J. Pharmacol. 2010, 160, 1595–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, H.L.; Wang, H.H.; Wu, C.Y.; Tung, W.H.; Yang, C.M. Lipoteichoic acid induces matrix metalloproteinase-9 expression via transactivation of PDGF receptors and NF-kappaB activation in rat brain astrocytes. Neurotox. Res. 2010, 17, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Su, E.J.; Fredriksson, L.; Geyer, M.; Folestad, E.; Cale, J.; Andrae, J.; Gao, Y.; Pietras, K.; Mann, K.; Yepes, M.; et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat. Med. 2008, 14, 731–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Hers, I.; Vincent, E.E.; Tavare, J.M. Akt signalling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef]
- Tallquist, M.; Kazlauskas, A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004, 15, 205–213. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Ahn, S.; Siddiqi, M.H.; Aceituno, V.C.; Simu, S.Y.; Yang, D.C. Suppression of MAPKs/NF-kappaB Activation Induces Intestinal Anti-Inflammatory Action of Ginsenoside Rf in HT-29 and RAW264.7 Cells. Immunol. Investig. 2016, 45, 439–449. [Google Scholar] [CrossRef]
- Ahn, S.-C.; Kim, G.-Y.; Kim, J.-H.; Baik, S.-W.; Han, M.-K.; Lee, H.-J.; Moon, D.-O.; Lee, C.-M.; Kang, J.-H.; Kim, B.-H. Epigallocatechin-3-gallate, constituent of green tea, suppresses the LPS-induced phenotypic and functional maturation of murine dendritic cells through inhibition of mitogen-activated protein kinases and NF-κB. Biochem. Biophys. Res. Commun. 2004, 313, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Mackman, N. LPS induction of gene expression in human monocytes. Cell. Signal. 2001, 13, 85–94. [Google Scholar] [CrossRef]
- Jou, T.C.; Jou, M.J.; Chen, J.Y.; Lee, S.Y. Properties of rat brain astrocytes in long-term culture. Taiwan Yi Xue Hui Za Zhi J. Formos. Med. Assoc. 1985, 84, 865–881. [Google Scholar]
- Wu, C.Y.; Hsieh, H.L.; Jou, M.J.; Yang, C.M. Involvement of p42/p44 MAPK, p38 MAPK, JNK and nuclear factor-kappa B in interleukin-1beta-induced matrix metalloproteinase-9 expression in rat brain astrocytes. J. Neurochem. 2004, 90, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, W.; Schulze, M.; Engels, C.; Klasmeier, E.; Pfeilschifter, J. Glucocorticoid-mediated suppression of cytokine-induced matrix metalloproteinase-9 expression in rat mesangial cells: Involvement of nuclear factor-kappaB and Ets transcription factors. Mol. Endocrinol. 2002, 16, 1752–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, H.L.; Wu, C.Y.; Yang, C.M. Bradykinin induces matrix metalloproteinase-9 expression and cell migration through a PKC-delta-dependent ERK/Elk-1 pathway in astrocytes. Glia 2008, 56, 619–632. [Google Scholar] [CrossRef]







 : Toll/IL-1 receptor (TIR) domain.
: Toll/IL-1 receptor (TIR) domain.
: Toll/IL-1 receptor (TIR) domain.
: Toll/IL-1 receptor (TIR) domain.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacological Inhibitor | Function |
|---|---|
| Actinomycin D | Transcription inhibitor: Binds DNA at the transcription initiation complex and prevents elongation of RNA chain by RNA polymerase. |
| AG1296 | Inhibitor of PDGFR: Inhibits the phosphorylation of autophosphorylation sites of the PDGFR. |
| Akt inhibitor VIII | Inhibitor of Akt: An allosteric inhibitor of Akt1 and Akt2 that less effectively blocks Akt3 activity. |
| Cycloheximide | Translation inhibitor: Inhibits translation elongation through binding to the E-site of the 60S ribosomal unit and interfering with deacetylated tRNA. |
| c-Src inhibitor II | Inhibitor of c-Src: A potent, selective, reversible, and ATP-competitive inhibitor of Src family tyrosine kinases. |
| LY294002 | Inhibitor of PI3K: Inhibits PI3K activity via competitive inhibition of an ATP binding site on the p85α subunit. |
| PF431396 | Inhibitor of Pyk2: An ATP-competitive inhibitor of PYK2/FAK (focal adhesion kinase). |
| p38 MAPK inhibitor VIII | Inhibitor of p38 MAPK: Inhibits p38α and p38β MAP kinase. |
| SP600125 | Inhibitor of JNK1-3: A reversible ATP-competitive inhibitor. |
| Tanshinone IIA | Inhibitor of AP-1: Inhibits AP-1 activity by suppressing jun-fos (members of the transcription factor AP-1 family)-DNA complex formation. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-C.; Lin, C.-C.; Hsiao, L.-D.; Kuo, J.-M.; Tseng, H.-C.; Yang, C.-M. Lipopolysaccharide-Induced Matrix Metalloproteinase-9 Expression Associated with Cell Migration in Rat Brain Astrocytes. Int. J. Mol. Sci. 2020, 21, 259. https://doi.org/10.3390/ijms21010259
Yang C-C, Lin C-C, Hsiao L-D, Kuo J-M, Tseng H-C, Yang C-M. Lipopolysaccharide-Induced Matrix Metalloproteinase-9 Expression Associated with Cell Migration in Rat Brain Astrocytes. International Journal of Molecular Sciences. 2020; 21(1):259. https://doi.org/10.3390/ijms21010259
Chicago/Turabian StyleYang, Chien-Chung, Chih-Chung Lin, Li-Der Hsiao, Jing-Ming Kuo, Hui-Ching Tseng, and Chuen-Mao Yang. 2020. "Lipopolysaccharide-Induced Matrix Metalloproteinase-9 Expression Associated with Cell Migration in Rat Brain Astrocytes" International Journal of Molecular Sciences 21, no. 1: 259. https://doi.org/10.3390/ijms21010259