M1 Macrophages Promote TRAIL Expression in Adipose Tissue-Derived Stem Cells, Which Suppresses Colitis-Associated Colon Cancer by Increasing Apoptosis of CD133+ Cancer Stem Cells and Decreasing M2 Macrophage Population

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Enhanced Expression of TRAIL in ASCs Cocultured with M1 Macrophages
2.2. Toxicity of TRAIL and CM in LoVo Cells
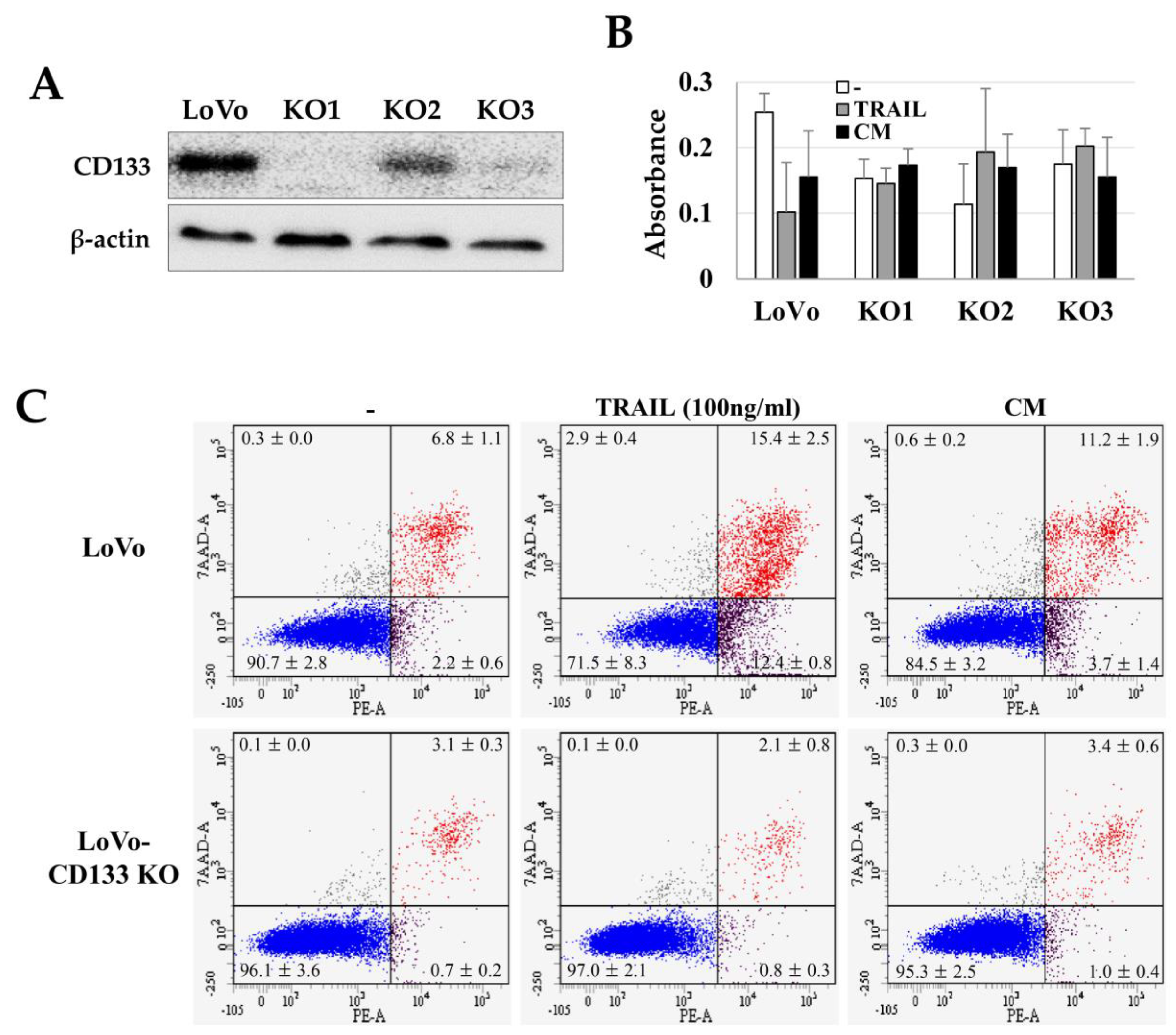
2.3. TRAIL Resistance of LoVo-CD133 KO Cells
2.4. Suppression of Colon Cancer Development and CD133 Expression by TRAIL-Expressing ASCs
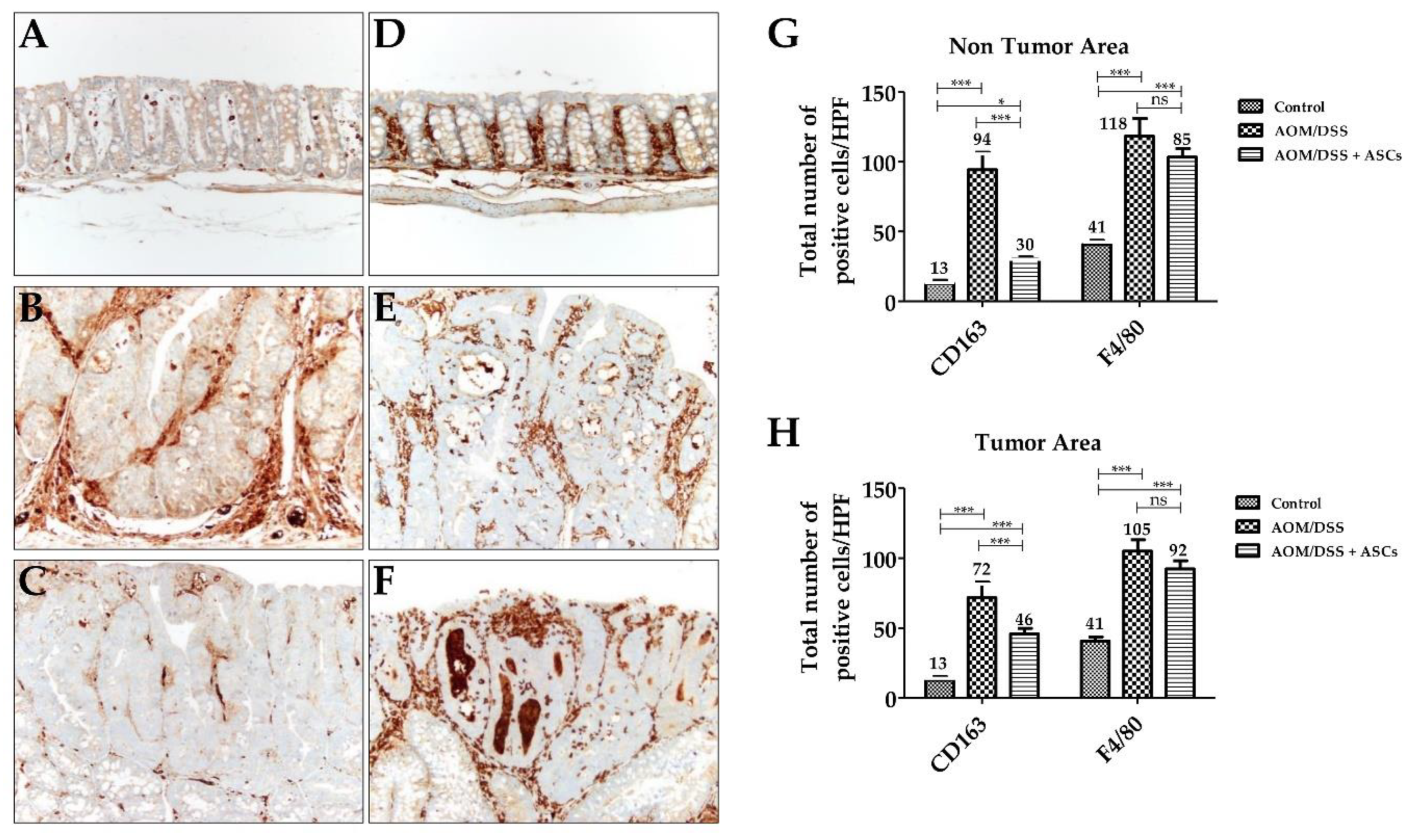
2.5. Decreasing M2 Macrophage Population Using TRAIL-Expressing ASCs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Next-Generation Sequencing (NGS)
4.3. Immunoblotting
4.4. Enzyme-Linked Immunosorbent Assay (ELISA)
4.5. Cytotoxicity Assay
4.6. Apoptosis Assay
4.7. Animal Study
4.8. Immunohistochemical (IHC) Staining
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Niidome, T.; Huang, L. Gene therapy progress and prospects: Nonviral vectors. Gene Ther. 2002, 9, 1647–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Huang, L. Nonviral gene therapy: Promises and challenges. Gene Ther. 2000, 7, 31–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Huang, L. Recent advances in nonviral vectors for gene delivery. Acc. Chem. Res. 2012, 45, 971–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Hyun, S.K.; Kim, H.B.; Kang, C.D.; Kim, S.H. Potential Role of CD133 Expression in the Susceptibility of Human Liver Cancer Stem-Like Cells to TRAIL. Oncol. Res. 2016, 24, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Fakiruddin, K.S.; Lim, M.N.; Nordin, N.; Rosli, R.; Zakaria, Z.; Abdullah, S. Targeting of CD133+ Cancer Stem Cells by Mesenchymal Stem Cell Expressing TRAIL Reveals a Prospective Role of Apoptotic Gene Regulation in Non-Small Cell Lung Cancer. Cancers 2019, 11, 1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Kessler, J.D.; Read, T.A.; Kaiser, C.; Corbeil, D.; Huttner, W.B.; Johnson, J.E.; Wechsler-Reya, R.J. Isolation of neural stem cells from the postnatal cerebellum. Nat. Neurosci. 2005, 8, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Clarke, I.D.; Hide, T.; Dirks, P.B. Cancer stem cells in nervous system tumors. Oncogene 2004, 23, 7267–7273. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef]
- Oshima, Y.; Suzuki, A.; Kawashimo, K.; Ishikawa, M.; Ohkohchi, N.; Taniguchi, H. Isolation of mouse pancreatic ductal progenitor cells expressing CD133 and c-Met by flow cytometric cell sorting. Gastroenterology 2007, 132, 720–732. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Miraglia, S.; Godfrey, W.; Yin, A.H.; Atkins, K.; Warnke, R.; Holden, J.T.; Bray, R.A.; Waller, E.K.; Buck, D.W. A novel five-transmembrane hematopoietic stem cell antigen: Isolation, characterization, and molecular cloning. Blood 1997, 90, 5013–5021. [Google Scholar] [CrossRef] [PubMed]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef]
- Wang, B.B.; Li, Z.J.; Zhang, F.F.; Hou, H.T.; Yu, J.K.; Li, F. Clinical significance of stem cell marker CD133 expression in colorectal cancer. Histol. Histopathol. 2016, 31, 299–306. [Google Scholar] [CrossRef]
- Zobalova, R.; McDermott, L.; Stantic, M.; Prokopova, K.; Dong, L.F.; Neuzil, J. CD133-positive cells are resistant to TRAIL due to up-regulation of FLIP. Biochem. Biophys. Res. Commun. 2008, 373, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Ruponen, M.; Honkakoski, P.; Ronkko, S.; Pelkonen, J.; Tammi, M.; Urtti, A. Extracellular and intracellular barriers in non-viral gene delivery. J. Control. Release 2003, 93, 213–217. [Google Scholar] [CrossRef]
- Park, S.J.; Park, W.; Na, K. Photo-activatable ternary complex based on a multifunctional shielding material for targeted shRNA delivery in cancer treatment. Biomaterials 2013, 34, 8991–8999. [Google Scholar] [CrossRef]
- Kim, K.S.; Park, W.; Na, K. Gadolinium-chelate nanoparticle entrapped human mesenchymal stem cell via photochemical internalization for cancer diagnosis. Biomaterials 2015, 36, 90–97. [Google Scholar] [CrossRef]
- Kim, S.M.; Lim, J.Y.; Park, S.I.; Jeong, C.H.; Oh, J.H.; Jeong, M.; Oh, W.; Park, S.H.; Sung, Y.C.; Jeun, S.S. Gene therapy using TRAIL-secreting human umbilical cord blood-derived mesenchymal stem cells against intracranial glioma. Cancer Res. 2008, 68, 9614–9623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komarova, S.; Roth, J.; Alvarez, R.; Curiel, D.T.; Pereboeva, L. Targeting of mesenchymal stem cells to ovarian tumors via an artificial receptor. J. Ovarian Res. 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selbo, P.K.; Weyergang, A.; Hogset, A.; Norum, O.J.; Berstad, M.B.; Vikdal, M.; Berg, K. Photochemical internalization provides time- and space-controlled endolysosomal escape of therapeutic molecules. J. Control. Release 2010, 148, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Oh, J.E.; Rhee, K.J.; Baik, S.K.; Kim, J.; Kang, S.J.; Sohn, J.H.; Choi, E.; Shin, H.C.; Kim, Y.M.; et al. Adipose tissue-derived mesenchymal stem cells cultured at high density express IFN-beta and suppress the growth of MCF-7 human breast cancer cells. Cancer Lett. 2014, 352, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Jung, P.Y.; Ryu, H.; Rhee, K.J.; Hwang, S.; Lee, C.G.; Gwon, S.Y.; Kim, J.; Kim, J.; Yoo, B.S.; Baik, S.K.; et al. Adipose tissue-derived mesenchymal stem cells cultured at high density express IFN-beta and TRAIL and suppress the growth of H460 human lung cancer cells. Cancer Lett. 2019, 440–441, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Byun, C.S.; Hwang, S.; Woo, S.H.; Kim, M.Y.; Lee, J.S.; Lee, J.I.; Kong, J.H.; Bae, K.S.; Park, I.H.; Kim, S.H.; et al. Adipose Tissue-Derived Mesenchymal Stem Cells Suppress Growth of Huh7 Hepatocellular Carcinoma Cells via Interferon (IFN)-beta-Mediated JAK/STAT1 Pathway in vitro. Int. J. Med. Sci. 2020, 17, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Pluddemann, A.; Martinez Estrada, F. Macrophage heterogeneity in tissues: Phenotypic diversity and functions. Immunol. Rev. 2014, 262, 36–55. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Sun, L.; Xu, F.; Liu, L.; Hu, F.; Song, D.; Hou, Z.; Wu, W.; Luo, X.; Wang, J.; et al. M2 Macrophage-Derived Exosomes Promote Cell Migration and Invasion in Colon Cancer. Cancer Res. 2019, 79, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.M.; Liu, K.; Liu, J.H.; Jiang, X.L.; Wang, X.L.; Chen, Y.Z.; Li, S.G.; Zou, H.; Pang, L.J.; Liu, C.X.; et al. CD163 as a marker of M2 macrophage, contribute to predicte aggressiveness and prognosis of Kazakh esophageal squamous cell carcinoma. Oncotarget 2017, 8, 21526–21538. [Google Scholar] [CrossRef] [Green Version]
- Shabo, I.; Olsson, H.; Elkarim, R.; Sun, X.F.; Svanvik, J. Macrophage Infiltration in Tumor Stroma is Related to Tumor Cell Expression of CD163 in Colorectal Cancer. Cancer Microenviron. 2014, 7, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.J.; Hsu, S.H. TRAIL-functionalized gold nanoparticles selectively trigger apoptosis in polarized macrophages. Nanotheranostics 2017, 1, 326–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Cho, M.Y.; Lee, S.; Jang, M.; Park, J.; Park, R. CRISPR-Cas9 mediated CD133 knockout inhibits colon cancer invasion through reduced epithelial-mesenchymal transition. PLoS ONE 2019, 14, e0220860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Na, K. The transfection efficiency of photosensitizer-induced gene delivery to human MSCs and internalization rates of EGFP and Runx2 genes. Biomaterials 2012, 33, 6485–6494. [Google Scholar] [CrossRef] [PubMed]
- Fakiruddin, K.S.; Ghazalli, N.; Lim, M.N.; Zakaria, Z.; Abdullah, S. Mesenchymal Stem Cell Expressing TRAIL as Targeted Therapy against Sensitised Tumour. Int. J. Mol. Sci. 2018, 19, 2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parang, B.; Barrett, C.W.; Williams, C.S. AOM/DSS Model of Colitis-Associated Cancer. Methods Mol. Biol. 2016, 1422, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Yamada, Y.; Sugie, S.; Mori, H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003, 94, 965–973. [Google Scholar] [CrossRef]
- De Robertis, M.; Massi, E.; Poeta, M.L.; Carotti, S.; Morini, S.; Cecchetelli, L.; Signori, E.; Fazio, V.M. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog 2011, 10, 9. [Google Scholar] [CrossRef]
- Chen, J.; Huang, X.F. The signal pathways in azoxymethane-induced colon cancer and preventive implications. Cancer Biol. Ther. 2009, 8, 1313–1317. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, R.; Kohno, H.; Sugie, S.; Nakagama, H.; Tanaka, T. Strain differences in the susceptibility to azoxymethane and dextran sodium sulfate-induced colon carcinogenesis in mice. Carcinogenesis 2006, 27, 162–169. [Google Scholar] [CrossRef]
- Egger, B.; Bajaj-Elliott, M.; MacDonald, T.T.; Inglin, R.; Eysselein, V.E.; Buchler, M.W. Characterisation of acute murine dextran sodium sulphate colitis: Cytokine profile and dose dependency. Digestion 2000, 62, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Melgar, S.; Karlsson, A.; Michaelsson, E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am. J. Phys. Gastrointest Liver Physiol. 2005, 288, G1328–G1338. [Google Scholar] [CrossRef] [PubMed]
- Ito, R.; Shin-Ya, M.; Kishida, T.; Urano, A.; Takada, R.; Sakagami, J.; Imanishi, J.; Kita, M.; Ueda, Y.; Iwakura, Y.; et al. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin. Exp. Immunol. 2006, 146, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Heinsbroek, S.E.; Gordon, S. The role of macrophages in inflammatory bowel diseases. Expert Rev. Mol. Med. 2009, 11, e14. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, J.; Saima, F.T.; Rhee, K.J.; Hwang, S.; Kim, M.Y.; Baik, S.K.; Eom, Y.W.; Kim, H.S. Adipose-derived stem cells ameliorate colitis by suppression of inflammasome formation and regulation of M1-macrophage population through prostaglandin E2. Biochem. Biophys Res. Commun. 2018, 498, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Fremond, C.M.; Yeremeev, V.; Nicolle, D.M.; Jacobs, M.; Quesniaux, V.F.; Ryffel, B. Fatal Mycobacterium tuberculosis infection despite adaptive immune response in the absence of MyD88. J. Clin. Invest. 2004, 114, 1790–1799. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, X.; Zheng, D.; Zhang, D.; Peng, X.; Zhang, X.; Ai, F.; Wang, X.; Ma, J.; Xiong, W.; et al. Dynamic changes and functions of macrophages and M1/M2 subpopulations during ulcerative colitis-associated carcinogenesis in an AOM/DSS mouse model. Mol. Med. Rep. 2015, 11, 2397–2406. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.R.; Pollock, K.; Hubel, A.; McKenna, D. Mesenchymal stem or stromal cells: A review of clinical applications and manufacturing practices. Transfusion 2014, 54, 1418–1437. [Google Scholar] [CrossRef]
- De Miguel, M.P.; Fuentes-Julian, S.; Blazquez-Martinez, A.; Pascual, C.Y.; Aller, M.A.; Arias, J.; Arnalich-Montiel, F. Immunosuppressive properties of mesenchymal stem cells: advances and applications. Curr. Mol. Med. 2012, 12, 574–591. [Google Scholar] [CrossRef]
- Qian, S.; Golubnitschaja, O.; Zhan, X. Chronic inflammation: Key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019, 10, 365–381. [Google Scholar] [CrossRef] [Green Version]
- Multhoff, G.; Molls, M.; Radons, J. Chronic inflammation in cancer development. Front. Immunol 2011, 2, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dite, P. Chronic inflammation is an important risk factor for the development of gastrointestinal cancer. Preface. Dig. Dis. 2010, 28, 573. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Inflammation and oncogenesis: A vicious connection. Curr. Opin. Genet. Dev. 2010, 20, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeNardo, D.G.; Johansson, M.; Coussens, L.M. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev. 2008, 27, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Masuda, J.; Shigehiro, T.; Matsumoto, T.; Satoh, A.; Mizutani, A.; Umemura, C.; Saito, S.; Kijihira, M.; Takayama, E.; Seno, A.; et al. Cytokine Expression and Macrophage Localization in Xenograft and Allograft Tumor Models Stimulated with Lipopolysaccharide. Int. J. Mol. Sci. 2018, 19, 1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Lee, M.R.; Choi, E.; Cho, M.Y. Clinicopathologic Significance of Survivin Expression in Relation to CD133 Expression in Surgically Resected Stage II or III Colorectal Cancer. J. Pathol. Transl. Med. 2017, 51, 17–23. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eom, Y.W.; Akter, R.; Li, W.; Lee, S.; Hwang, S.; Kim, J.; Cho, M.-Y. M1 Macrophages Promote TRAIL Expression in Adipose Tissue-Derived Stem Cells, Which Suppresses Colitis-Associated Colon Cancer by Increasing Apoptosis of CD133+ Cancer Stem Cells and Decreasing M2 Macrophage Population. Int. J. Mol. Sci. 2020, 21, 3887. https://doi.org/10.3390/ijms21113887
Eom YW, Akter R, Li W, Lee S, Hwang S, Kim J, Cho M-Y. M1 Macrophages Promote TRAIL Expression in Adipose Tissue-Derived Stem Cells, Which Suppresses Colitis-Associated Colon Cancer by Increasing Apoptosis of CD133+ Cancer Stem Cells and Decreasing M2 Macrophage Population. International Journal of Molecular Sciences. 2020; 21(11):3887. https://doi.org/10.3390/ijms21113887
Chicago/Turabian StyleEom, Young Woo, Rokeya Akter, Wanlu Li, Suji Lee, Soonjae Hwang, Jiye Kim, and Mee-Yon Cho. 2020. "M1 Macrophages Promote TRAIL Expression in Adipose Tissue-Derived Stem Cells, Which Suppresses Colitis-Associated Colon Cancer by Increasing Apoptosis of CD133+ Cancer Stem Cells and Decreasing M2 Macrophage Population" International Journal of Molecular Sciences 21, no. 11: 3887. https://doi.org/10.3390/ijms21113887