Podocytes—The Most Vulnerable Renal Cells in Preeclampsia

 , , , ,
, , , , {kind=link}
{kind=link}
Abstract
:1. Preeclampsia
2. Glomerular Lesions Secondary to Preeclampsia
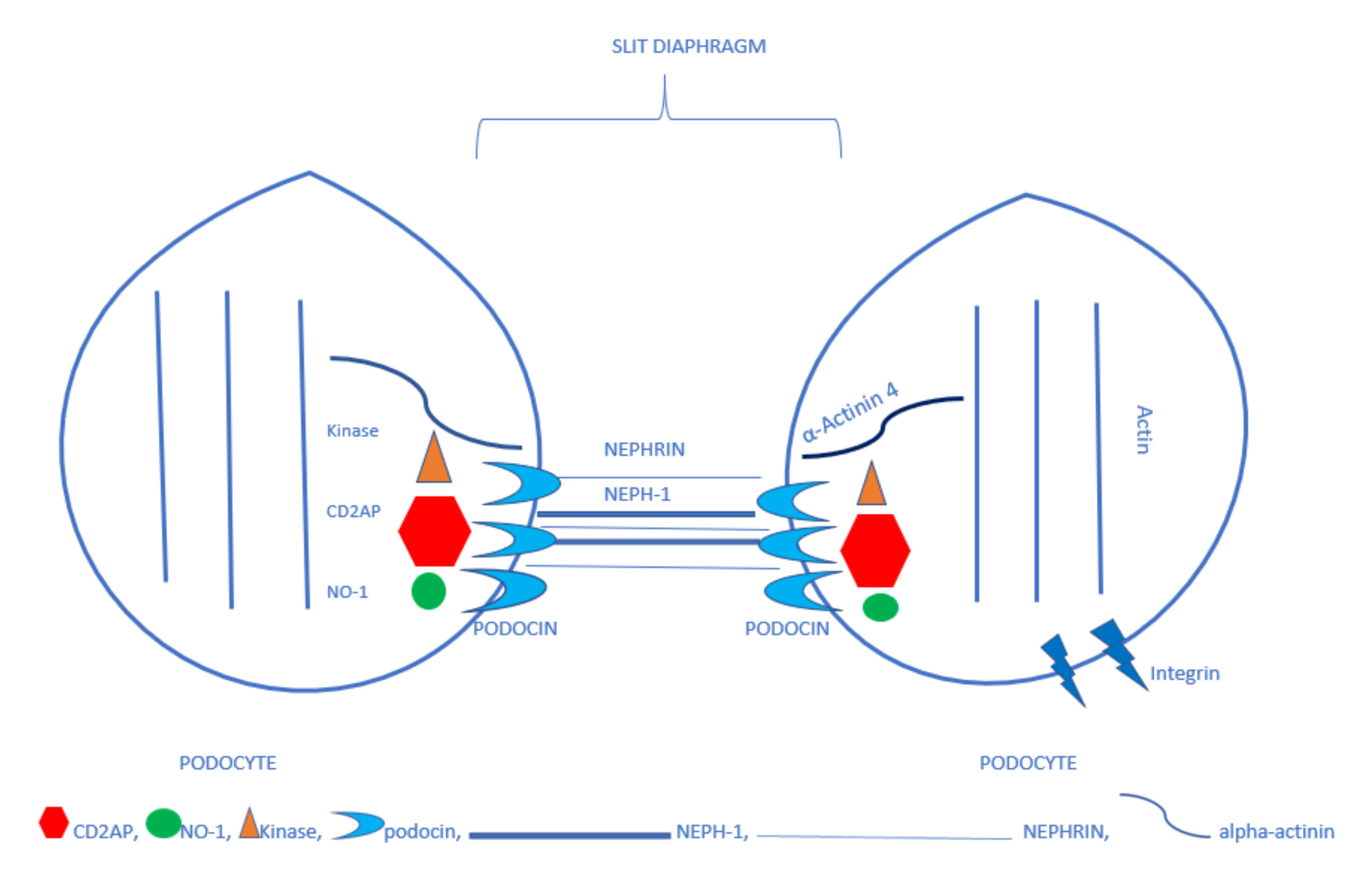
3. Podocytes
4. Urinary Excretion of Podocytes and Their Proteins Secondary to Preeclampsia
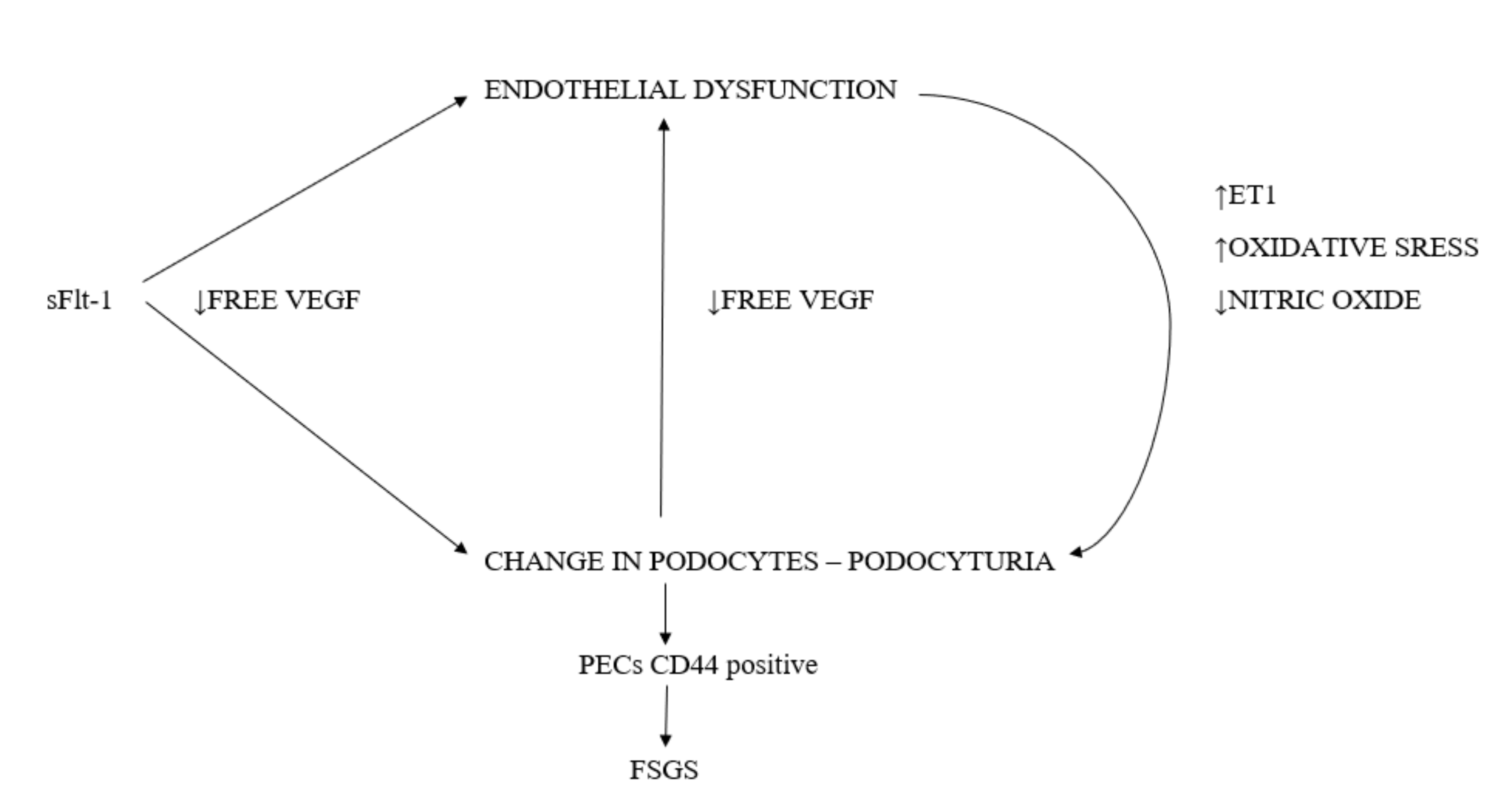
5. The Podocyte Damage Mechanism in Preeclampsia
Free VEGF Deficiency
6. Endothelial Damage-Related Disorders
6.1. Nitric Oxide (NO) Deficiency
6.2. Endothelin-1
6.3. Oxidative Stress
7. Preeclampsia and the Risk of Developing End-Stage Renal Disease (ESRD)
8. The Mechanism behind Focal Segmental Glomerulosclerosis (FSGS)
9. Conclusions
Funding
Conflicts of Interest
References
- Tomimatsu, T.; Mimura, K.; Endo, M.; Kumasawa, K.; Kimura, T. Pathophysiology of Preeclampsia: An Angiogenic Imbalance and Long-Lasting Systemic Vascular Dysfunction. Hypertens. Res. 2017, 40, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Hussein, W.; Lafayette, R.A. Renal Function in Normal and Disordered Pregnancy. Curr. Opin. Nephrol. Hypertens. 2014, 23, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, M.J.; Hutchinson, K.; Mathieson, P.W.; Witherden, I.R.; Saleem, M.A.; Hunter, M. Human podocytes possess a stretch-sensitive, Ca2+-activated K+ channel: Potential implications for the control of glomerular filtration. J. Am. Soc. Nephrol. 2004, 15, 2981–2987. [Google Scholar] [CrossRef] [Green Version]
- Mundel, P.; Kriz, W. Structure and function of podocytes: An update. Anat. Embryol. 1995, 192, 385–397. [Google Scholar] [CrossRef]
- Asanuma, K.; Shirato, I.; Ishidoh, K.; Kominami, E.; Tomino, Y. Selective modulation of the secretion of proteinases and their inhibitors by growth factors in cultured differentiated podocytes. Kidney Int. 2002, 62, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Armaly, Z.; Jadaon, J.E.; Jabbour, A.; Abassi, Z.A. Preeclampsia: Novel Mechanisms and Potential Therapeutic Approaches. Front. Physiol. 2018, 259, 973. [Google Scholar] [CrossRef] [Green Version]
- Humphries, J.D.; Wang, P.; Streuli, C.; Geiger, B.; Humphries, M.J.; Ballestrem, C. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J. Cell Biol. 2007, 179, 1043–1057. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, K.; Kurihara, H.; Sakai, T. Actin filament organization of foot processes in rat podocytes. J. Histochem. Cytochem. 2003, 51, 1589–1600. [Google Scholar] [CrossRef] [Green Version]
- Philippe, A.; Nevo, F.; Esquivel, E.L.; Reklaityte, D.; Gribouval, O.; Tete, M.J.; Loirat, C.; Dantal, J.; Fischbach, M.; Pouteil-Noble, C.; et al. Nephrin mutations can cause childhood-onset steroid-resistant nephrotic syndrome. J. Am. Soc. Nephrol. 2008, 19, 1871–1878. [Google Scholar] [CrossRef] [Green Version]
- Garg, P.; Verma, R.; Nihalani, D.; Johnstone, D.B.; Holzman, L.B. Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol. Cell Biol. 2007, 27, 8698–8712. [Google Scholar] [CrossRef] [Green Version]
- Jalanko, H. Pathogenesis of proteinuria: Lessons learned from nephrin and podocin. Pediatr. Nephrol. 2003, 18, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Grunkemeyer, J.A.; Kwoh, C.; Huber, T.B.; Shaw, A.S. CD2-associated Protein (CD2AP) Expression in Podocytes Rescues Lethality of CD2AP Deficiency. J. Biol. Chem. 2005, 19, 29677–29681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grahammer, F.; Schell, C.; Huber, T.B. The podocyte slit diaphragm—From a thin grey line to a complex signalling hub. Nat. Rev. Nephrol. 2013, 9, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.M.; Kim, S.H.; North, K.N.; Rennke, H.; Correia, L.A.; Tong, H.Q.; Mathis, B.J.; Rodríguez-Pérez, J.C.; Allen, P.G.; Beggs, A.H.; et al. Mutations in ACTN4, Encoding alpha-actinin-4, Cause Familial Focal Segmental Glomerulosclerosis. Nat. Genet. 2000, 24, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Meyrier, A. Mechanisms of Disease: Focal Segmental Glomerulosclerosis. Nat. Clin. Pract. Nephrol. 2005, 1, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Craici, I.M.; Wagner, S.J.; Bailey, K.R.; Fitz-Gibbon, P.D.; Wood-Wentz, C.M.; Turner, S.T.; Hayman, S.R.; White, W.M.; Brost, B.C.; Rose, C.H.; et al. Podocyturia predates proteinuria and clinical features of preeclampsia: Longitudinal prospective study. Hypertension 2013, 61, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Liapis, H.; Romagnani, P.; Anders, H.J. New Insights into the Pathology of Podocyte Loss: Mitotic catastrophe. Am. J. Pathol. 2013, 183, 1364–1374. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir, F.; Tayyar, A.T.; Acmaz, G.; Aksoy, H.; Erturk, G.; Muhtaroglu, S.; Tayyar, M. Comparison of blood and urine nephrin levels in preeclampsia and intrauterine growth retardation. Pak. J. Med. Sci. 2016, 32, 40–43. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gu, Y.; Loyd, S.; Jia, X.; Groome, L.J. Increased urinary levels of podocyte glycoproteins, matrix metallopeptidases, inflammatory cytokines, and kidney injury biomarkers in women with preeclampsia. Am. J. Physiol. Renal Physiol. 2015, 309, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.J.; Cho, H.Y.; Cho, S.; Kim, Y.H.; Jeon, J.D.; Kim, Y.J.; Lee, S.; Park, J.; Kim, H.Y.; Park, Y.W.; et al. The Level of Serum and Urinary Nephrin in Normal Pregnancy and Pregnancy with Subsequent Preeclampsia. Yonsei Med. J. 2017, 58, 401–406. [Google Scholar] [CrossRef]
- Garovic, V.D.; Wagner, S.J.; Turner, S.T.; Rosenthal, D.W.; Watson, W.J.; Brost, B.C.; Rose, C.H.; Gavrilova, L.; Craigo, P.; Bailey, K.R.; et al. Urinary podocyte excretion as a marker for preeclampsia. Am. J. Obstet. Gynecol. 2007, 196, 320. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, S.; Gu, Y.; Lewis, D.F. Loss of slit protein nephrin is associated with reduced antioxidant superoxide dismutase expression in podocytes shed from women with preeclampsia. Physiol. Rep. 2018, 6, 13785. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, A.; Ryba, M.; Wartacz, J.; Czyżewska-Buczyńska, A.; Hruby, Z.; Witkiewicz, W. Podocytes in Urine, a Novel Biomarker of Preeclampsia? Adv. Clin. Exp. Med. 2013, 22, 145–149. [Google Scholar] [PubMed]
- Hauser, P.V.; Collino, F.; Bussolati, B.; Camussi, G. Nephrin and endothelial injury. Curr. Opin. Nephrol. Hypertens. 2009, 18, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Collino, F.; Bussolati, B.; Gerbaudo, E.; Marozio, L.; Pelissetto, S.; Benedetto, C.; Camussi, G. Preeclamptic sera induce nephrin shedding from podocytes through endothelin-1 release by endothelial glomerular cells. Am. J. Physiol. Renal Physiol. 2008, 294, 1185–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerley, R.N.; McCarthy, C. Biomarkers of Glomerular Dysfunction in Pre-Eclampsia—A Systematic Review. Pregnancy Hypertens. 2018, 14, 265–272. [Google Scholar] [CrossRef]
- Martineau, T.; Boutin, M.; Côté, A.M.; Maranda, B.; Bichet, D.G.; Auray-Blais, C. Tandem Mass Spectrometry Analysis of Urinary Podocalyxin and Podocin in the Investigation of Podocyturia in Women with Preeclampsia and Fabry Disease Patients. Clin. Chim. Acta 2019, 495, 67–75. [Google Scholar] [CrossRef]
- Gilani, S.I.; Anderson, U.D.; Jayachandran, M.; Weissgerber, T.L.; Zand, L.; White, W.M.; Milic, N.; Gonzalez Suarez, M.L.; Vallapureddy, R.R.; Nääv, A.; et al. Urinary Extracellular Vesicles of Podocyte Origin and Renal Injury in Preeclampsia. J. Am. Soc. Nephrol. 2017, 28, 3363–3372. [Google Scholar] [CrossRef]
- Baijnath, S.; Murugesan, S.; Mackraj, I.; Gathiram, P.; Moodley, J. The effects of sildenafil citrate on urinary podocin and nephrin mRNA expression in an L-NAME model of pre-eclampsia. Mol. Cell. Biochem. 2016, 427, 59–67. [Google Scholar] [CrossRef]
- Henao, D.E.; Arias, L.F.; Mathieson Ni, L.; Welsh, G.I.; Bueno, J.C.; Agudelo, B.; Cadavid, A.P.; Saleem, M.A. Preeclamptic Sera Directly Induce Slit-Diaphragm Protein Redistribution and Alter Podocyte Barrier-Forming Capacity. Nephron. Exp. Nephrol. 2008, 110, 73–81. [Google Scholar] [CrossRef]
- Guan, F.; Villegas, G.; Teichman, J.; Mundel, P.; Tufro, A. Autocrine VEGF-A System in Podocytes Regulates Podocin and Its Interaction with CD2AP. Am. J. Physiol. Renal Physiol. 2006, 291, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Quaggin, S.E. The Role of VEGF-A in Glomerular Development and Function. Curr. Opin. Nephrol. Hypertens. 2004, 13, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.J.; Craici, I.M.; Grande, J.P.; Garovic, V.D. From Placenta to Podocyte: Vascular and Podocyte Pathophysiology in Preeclampsia. Clin. Nephrol. 2012, 78, 241–249. [Google Scholar] [CrossRef]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Invest. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tufro, A.; Veron, D. VEGF and Podocytes in Diabetic Nephropathy. Semin. Nephrol. 2012, 32, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller-Deile, J.; Bröcker, V.; Grünwald, V.; Hiss, M.; Bertram, A.; Kubicka, S.; Ganser, A.; Haller, H.; Schiffer, M. Renal side effects of VEGF-blocking therapy. NDT Plus 2010, 3, 172–175. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Kwiatkowska, E.; Torbe, A. The role of disordered angiogenesis tissue markers (sflt-1, Plgf) in present day diagnosis of preeclampsia. Ginekol. Pol. 2019, 90, 173–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Trivedi, S.S.; Bhattacharjee, J. Oxidative Stress and eNOS (Glu298Asp) Gene Polymorphism in Preeclampsia in Indian Population. Mol. Cell Biochem. 2011, 353, 189–193. [Google Scholar] [CrossRef]
- Saleh, L.; Verdonk, K.; Visser, W.; Meiracker, A.H.; Danser, A.H.J. The Emerging Role of endothelin-1 in the Pathogenesis of Pre-Eclampsia. Ther. Adv. Cardiovasc. Dis. 2016, 10, 282–293. [Google Scholar] [CrossRef]
- Murphy, S.R.; LaMarca, B.B.D.; Cockrell, K.; Granger, J.P. Role of Endothelin in Mediating Soluble Fms-Like Tyrosine Kinase 1-induced Hypertension in Pregnant Rats. Hypertension 2010, 55, 394–398. [Google Scholar] [CrossRef] [Green Version]
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Gu, X.; Groome, L.J.; Wang, Y. Decreased Nephrin and GLEPP-1, But Increased VEGF, Flt-1, and Nitrotyrosine, Expressions in Kidney Tissue Sections from Women with Preeclampsia. Reprod. Sci. 2009, 16, 970–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vikse, B.E.; Irgens, M.; Leivestad, T.; Skjaerven, R.; Iversen, B.M. Preeclampsia and the Risk of End-Stage Renal Disease. N. Engl. J. Med. 2008, 359, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.C.; Pell, J.P.; Walsh, D. Pregnancy Complications and Maternal Risk of Ischaemic Heart Disease: A Retrospective Cohort Study of 129,290 Births. Lancet 2001, 357, 2002–2006. [Google Scholar] [CrossRef]
- Bar, J.; Kaplan, B.; Wittenberg, C.; Erman, A.; Boner, G.; Ben-Rafael, Z.; Hod, M. Microalbuminuria After Pregnancy Complicated by Pre-Eclampsia. Nephrol. Dial. Transplant. 1999, 14, 1129–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khashan, A.S.; Evans, M.; Kublickas, M.; McCarthy, F.P.; Kenny, L.C.; Stenvinkel, P.; Fitzgerald, T.; Kublickiene, K. Preeclampsia and Risk of End Stage Kidney Disease: A Swedish Nationwide Cohort Study. PLoS Med. 2019, 16, 1002875. [Google Scholar] [CrossRef] [Green Version]
- Webster, P.; Webster, L.M.; Cook, H.T.; Horsfield, C.; Seed, P.T.; Vaz, R.; Santos, C.; Lydon, I.; Homsy, M.; Lightstone, L.; et al. A Multicenter Cohort Study of Histologic Findings and Long-Term Outcomes of Kidney Disease in Women Who Have Been Pregnant. Clin. J. Am. Soc. Nephrol. 2017, 12, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Kida, H.; Takeda, S.; Yokoyama, H.; Tomosugi, N.; Abe, T.; Hattori, N. Focal glomerular sclerosis in pre-eclampsia. Clin. Nephrol. 1985, 24, 221–227. [Google Scholar] [PubMed]
- Nochy, D.; Hinglais, N.; Jacquot, C.; Gaudry, C.; Remy, P.; Bariety, J. De novo focal glomerular sclerosis in preeclampsia. Clin. Nephrol. 1986, 25, 116–121. [Google Scholar]
- Nagai, Y.; Arai, H.; Washizawa, Y.; Ger, Y.; Tanaka, M.; Maeda, M.; Kawamura, S. FSGS-like lesions in pre-eclampsia. Clin. Nephrol. 1991, 36, 134–140. [Google Scholar]
- Wharram, B.L.; Goyal, M.; Wiggins, J.E.; Sanden, S.K.; Hussain, S.; Filipiak, W.E.; Saunders, T.L.; Dysko, R.C.; Kohno, K.; Holzman, L.B.; et al. Podocyte Depletion Causes Glomerulosclerosis: Diphtheria Toxin-Induced Podocyte Depletion in Rats Expressing Human Diphtheria Toxin Receptor Transgene. J. Am. Soc. Nephrol. 2005, 16, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, A.; Wickman, L.T.; Venkatareddy, M.P.; Sato, Y.; Chowdhury, M.A.; Wang, S.Q.; Shedden, K.A.; Dysko, R.C.; Wiggins, J.E.; Wiggins, R.C. Angiotensin II-dependent Persistent Podocyte Loss From Destabilized Glomeruli Causes Progression of End Stage Kidney Disease. Kidney Int. 2012, 81, 40–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsusaka, T.; Xin, J.; Niwa, S.; Kobayashi, K.; Akatsuka, A.; Hashizume Wang, Q.C.; Pastan, I.; Fogo, A.G.; Ichikawa, L. Genetic Engineering of Glomerular Sclerosis in the Mouse via Control of Onset and Severity of Podocyte-Specific Injury. J. Am. Soc. Nephrol. 2005, 16, 1013–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeets, B.; Kuppe, C.; Sicking, E.M.; Fuss, A.; Jirak, P.; Kuppevelt, T.H.; Endlich, K.; Wetzels, J.F.M.; Gröne, H.J.; Floege, J.; et al. Parietal Epithelial Cells Participate in the Formation of Sclerotic Lesions in Focal Segmental Glomerulosclerosis. J. Am. Soc. Nephrol. 2011, 22, 1262–1274. [Google Scholar] [CrossRef] [Green Version]
- Saritas, T.; Moeller, M.J. Glomerular Disease: Pre-Eclampsia, Podocyturia and the Role of Parietal Epithelial Cells. Nat. Rev. Nephrol. 2014, 10, 615–616. [Google Scholar] [CrossRef]
- Jefferson, J.A.; Shankland, S.J. The Pathogenesis of Focal Segmental Glomerulosclerosis. Adv. Chronic Kidney Dis. 2014, 21, 408–416. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.M.; Ohse, T.; Krofft, R.D.; Wu, J.S.; Eddy, A.A.; Pippin, J.W.; Shankland, S.J. Albumin-induced Apoptosis of Glomerular Parietal Epithelial Cells Is Modulated by Extracellular Signal-Regulated Kinase 1/2. Nephrol. Dial. Transplant. 2012, 27, 1330–1343. [Google Scholar] [CrossRef] [Green Version]
- Erkan, E.; Garcia, C.D.; Patterson, L.T.; Mishra, J.; Mitsnefes, M.; Kaskel, F.J.; Devarajan, P. Induction of Renal Tubular Cell Apoptosis in Focal Segmental Glomerulosclerosis: Roles of Proteinuria and Fas-Dependent Pathways. J. Am. Soc. Nephrol. 2005, 16, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Matsusaka, T.; Sandgren, E.; Shintani, A.; Kon, V.; Pastan, I.; Fogo, A.G.; Ichikawa, I. Podocyte Injury Damages Other Podocytes. J. Am. Soc. Nephrol. 2011, 22, 1275–1285. [Google Scholar] [CrossRef]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF Inhibition and Renal Thrombotic Microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwiatkowska, E.; Stefańska, K.; Zieliński, M.; Sakowska, J.; Jankowiak, M.; Trzonkowski, P.; Marek-Trzonkowska, N.; Kwiatkowski, S. Podocytes—The Most Vulnerable Renal Cells in Preeclampsia. Int. J. Mol. Sci. 2020, 21, 5051. https://doi.org/10.3390/ijms21145051
Kwiatkowska E, Stefańska K, Zieliński M, Sakowska J, Jankowiak M, Trzonkowski P, Marek-Trzonkowska N, Kwiatkowski S. Podocytes—The Most Vulnerable Renal Cells in Preeclampsia. International Journal of Molecular Sciences. 2020; 21(14):5051. https://doi.org/10.3390/ijms21145051
Chicago/Turabian StyleKwiatkowska, Ewa, Katarzyna Stefańska, Maciej Zieliński, Justyna Sakowska, Martyna Jankowiak, Piotr Trzonkowski, Natalia Marek-Trzonkowska, and Sebastian Kwiatkowski. 2020. "Podocytes—The Most Vulnerable Renal Cells in Preeclampsia" International Journal of Molecular Sciences 21, no. 14: 5051. https://doi.org/10.3390/ijms21145051