Characterization of Fluid Biomarkers Reveals Lysosome Dysfunction and Neurodegeneration in Neuronopathic MPS II Patients


 , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Results
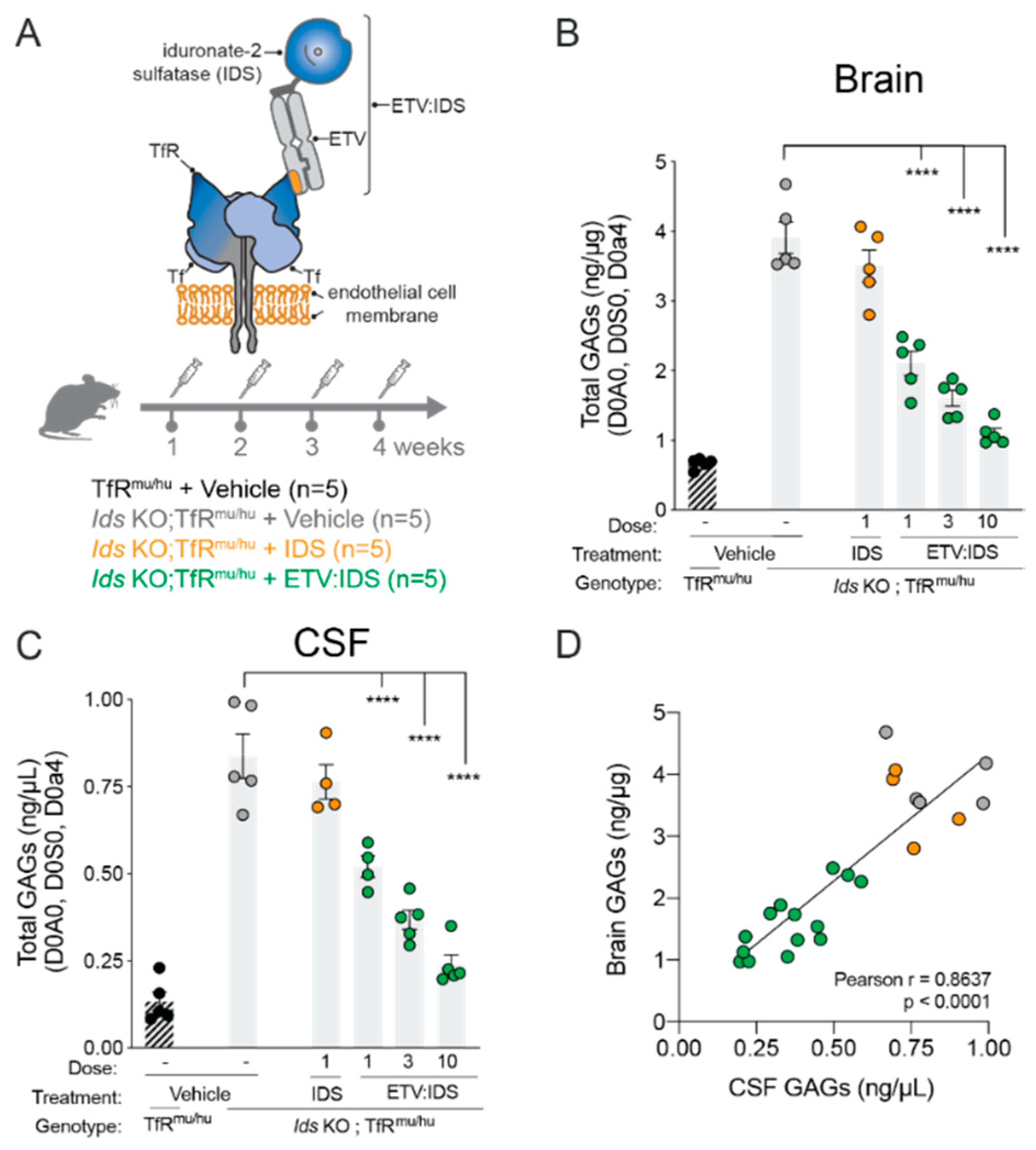
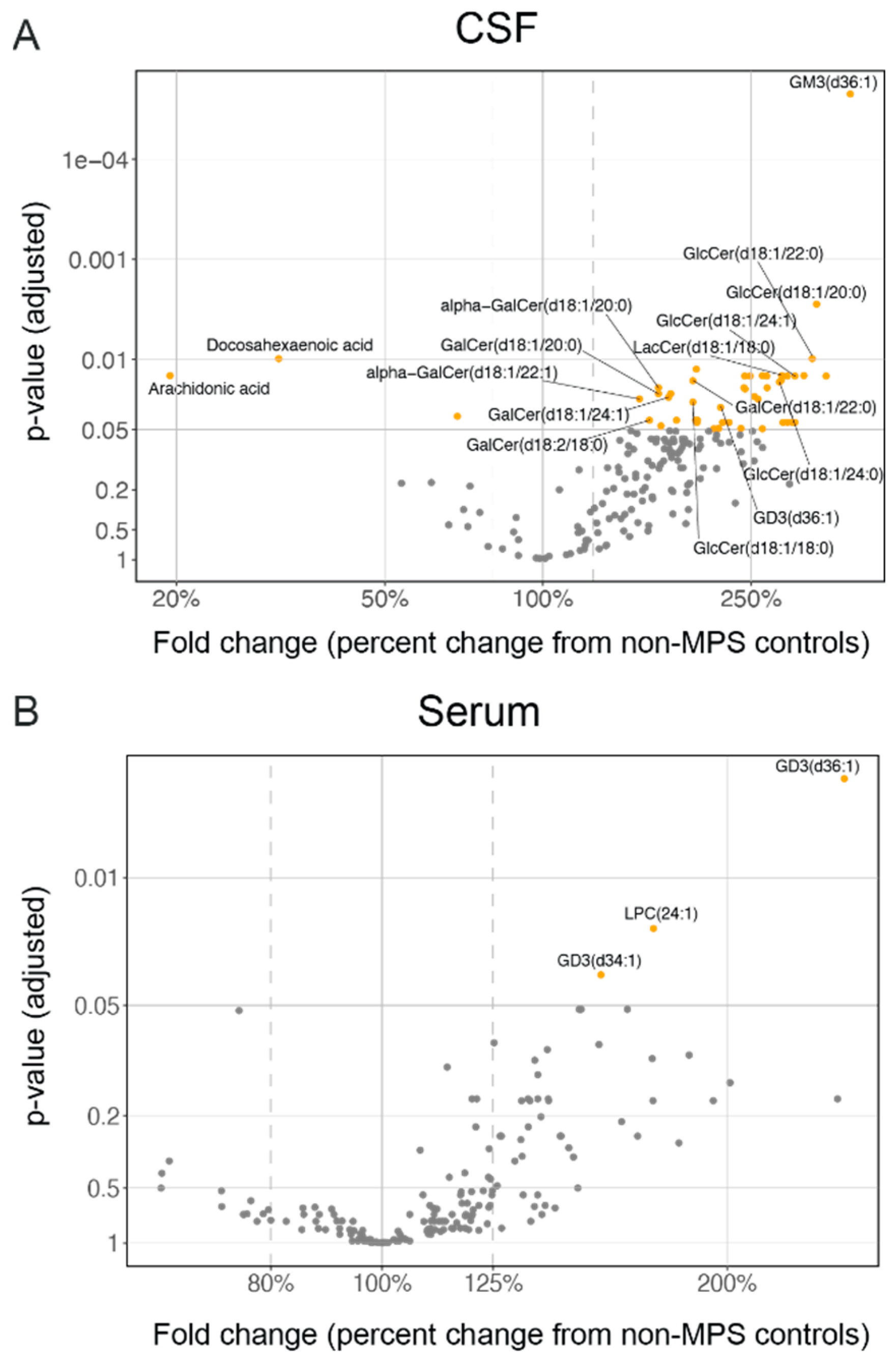
2.1. High Degree of Correlation between Brain and CSF Glycosaminoglycan (GAG) as Well as Secondary Storage Substrates Such as Gangliosides (GM3)
2.2. nMPS II Patients on Enzyme Replacement Therapy (ERT) or Hematopoietic Stem Cell Transplant (HSCT) Show Elevated Levels of HS and DS in CSF and Serum
2.3. Lysosomal Lipids Are Elevated in CSF and Serum of MPS II Individuals
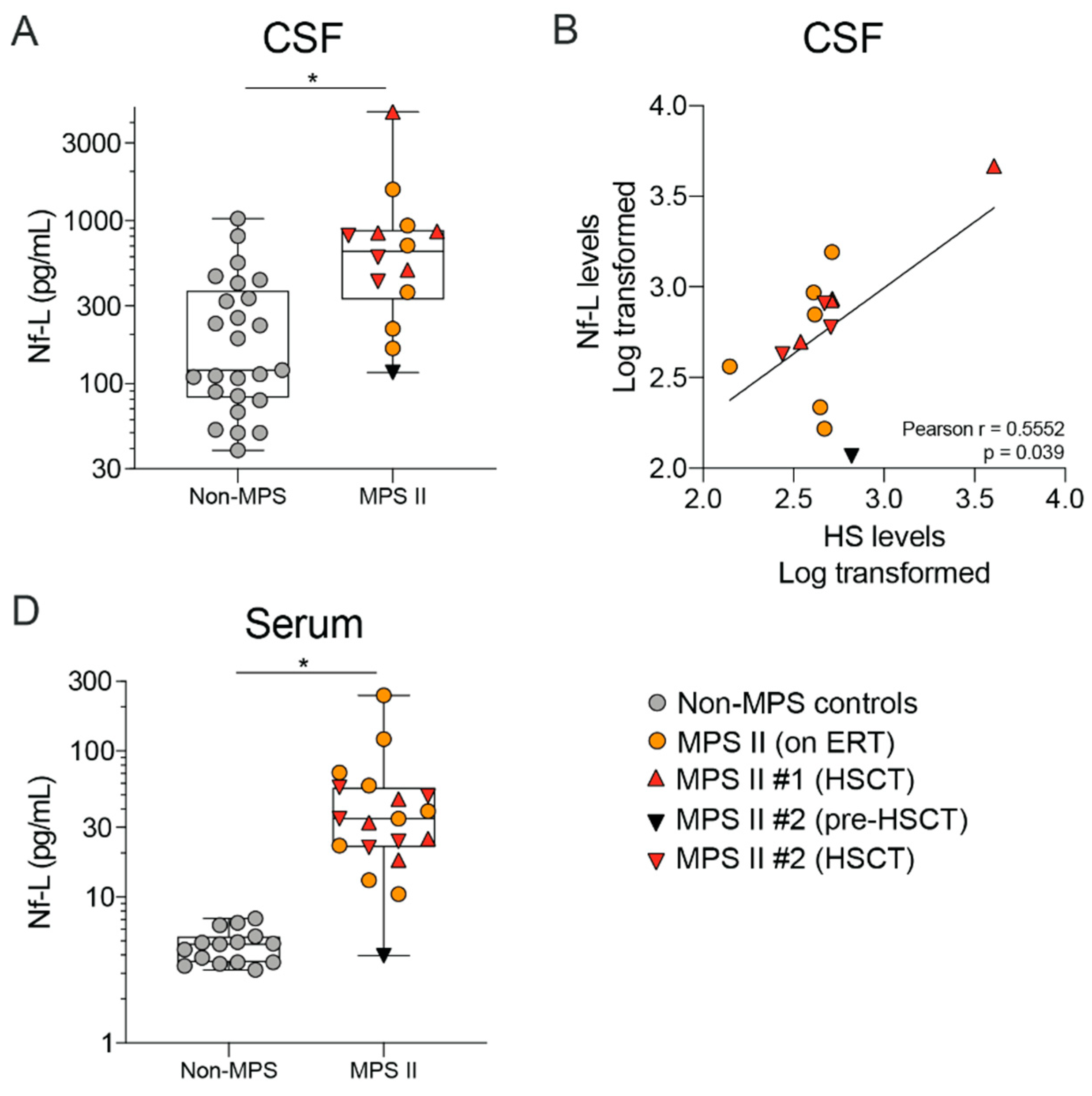
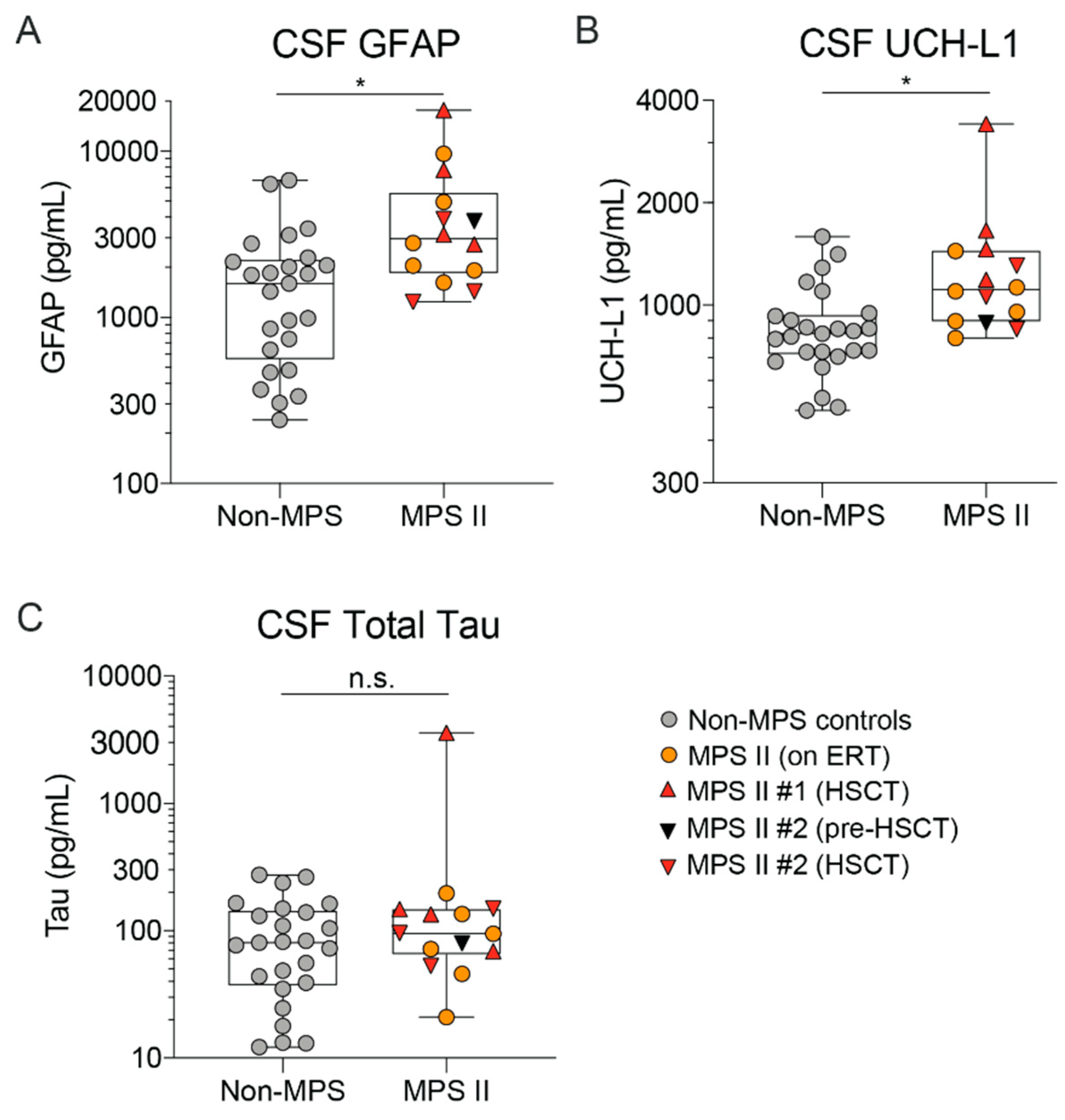
2.4. MPS II Patient CSF and Serum Show Altered Levels of Neurodegeneration Markers Such as Nf-L
3. Discussion
4. Materials and Methods
4.1. Collection of MPS II Patient and Non-MPS Control Samples
4.2. Expression, Purification and Characterization of Recombinant Human ETV:IDS and IDS
4.3. Animal Care, Generation of Mouse Strains and Dosing Methods
4.4. Preclinical Tissue and Fluid Processing for Downstream Biomarker Analysis
4.5. Nf-L and Neurology 4-Plex Analyte Analysis of Human CSF and Serum
4.6. HS and DS Analysis in Human CSF and Serum
4.7. LC-MS Assay for BMP, Glucosylceramides and Gangliosides
4.8. Exploratory Analysis of Lipids in Human CSF and Serum
4.9. Statistical Analysis of Quantified Endpoints and Exploratory Lipidomics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BBB | Blood-Brain Barrier |
| IDS | Iduronate 2-Sulfatase (Elaprase™) |
| GAG | Glycosaminoglycan |
| LC-MS/MS | Liquid Chromatography-Tandem Mass Spectrometry |
| HS | Heparan Sulfate |
| DS | Dermatan Sulfate |
| CSF | Cerebrospinal Fluid |
| TfR | Transferrin Receptor |
| MPS II | Mucopolysaccharidosis II |
| ETV | Enzyme Transport Vehicle |
| D0A0 | 4,5 unsaturated uronic acid residue-N-acetyl glucosamine, HS ∆UA-GluNAc |
| D0S0 | 4,5 unsaturated uronic acid residue-glucosamine-N-sulfate, ∆UA-GluNS |
| D0a4 | 4,5 unsaturated uronic acid residue-N-acetyl galactosamine, 4-O-sulfate, ∆UA-GalNAc4S |
| HSCT | Hematopoietic Stem Cell Transplant |
| ERT | Enzyme Replacement Therapy |
| Nf-L | Neurofilament Light Chain |
| TfRmu/hu KI | Chimeric human/mouse Transferrin Receptor Knock-In mouse |
References
- Viana, G.M.; Priestman, D.A.; Platt, F.M.; Khan, S.; Tomatsu, S.; Pshezhetsky, A.V. Brain Pathology in Mucopolysaccharidoses (MPS) Patients with Neurological Forms. J. Clin. Med. 2020, 9, 396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R.; Beck, M.; Eng, C.; Giugliani, R.; Harmatz, P.; Muñoz, V.; Muenzer, J. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008, 121, e377–e386. [Google Scholar] [CrossRef] [PubMed]
- D’Avanzo, F.; Rigon, L.; Zanetti, A.; Tomanin, R. Mucopolysaccharidosis Type II: One Hundred Years of Research, Diagnosis, and Treatment. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [Green Version]
- Lampe, C.; Atherton, A.; Burton, B.K.; Descartes, M.; Giugliani, R.; Horovitz, D.D.; Kyosen, S.O.; Magalhães, T.S.; Martins, A.M.; Mendelsohn, N.J.; et al. Enzyme Replacement Therapy in Mucopolysaccharidosis II Patients Under 1 Year of Age. JIMD Rep. 2014, 14, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Hendriksz, C.J.; Muenzer, J.; Vanderver, A.; Davis, J.M.; Burton, B.K.; Mendelsohn, N.J.; Wang, N.; Pan, L.; Pano, A.; Barbier, A.J. Levels of glycosaminoglycans in the cerebrospinal fluid of healthy young adults, surrogate-normal children, and Hunter syndrome patients with and without cognitive impairment. Mol. Genet. Metab. Rep. 2015, 5, 103–106. [Google Scholar] [CrossRef]
- Hendriksz, C.; Muenzer, J.; Burton, B.; Pan, L.; Wang, N.; Naimy, H.; Pano, A.; Barbier, A. A Cerebrospinal Fluid Collection Study in Pediatric and Adult Patients With Hunter Syndrome. J. Inborn Errors Metab. Screen. 2015, 1. [Google Scholar] [CrossRef]
- Walkley, S.U. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Boil. 2004, 15, 433–444. [Google Scholar] [CrossRef]
- Walkley, S.U.; Vanier, M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta 2009, 1793, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantopoulos, G.; Iqbal, K.; Dekaban, A.S. Mucopolysaccharidosis Types IH, IS, II and IIIA: Glycosaminoglycans and Lipids of Isolated Brain Cells and Other Fractions from Autopsied Tissues. J. Neurochem. 1980, 34, 1399–1411. [Google Scholar] [CrossRef]
- Saville, J.T.; Thai, H.N.; Lehmann, R.J.; Derrick-Roberts, A.L.K.; Fuller, M. Subregional brain distribution of simple and complex glycosphingolipids in the mucopolysaccharidosis type I (Hurler syndrome) mouse: Impact of diet. J. Neurochem. 2017, 141, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Polito, V.A.; Cosma, M.P. IDS crossing of the blood-brain barrier corrects CNS defects in MPSII mice. Am. J. Hum. Genet. 2009, 85, 296–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef] [PubMed]
- Fujitsuka, H.; Sawamoto, K.; Peracha, H.; Mason, R.W.; Mackenzie, W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Biomarkers in patients with mucopolysaccharidosis type II and IV. Mol. Genet. Metab. Rep. 2019, 19, 100455. [Google Scholar] [CrossRef]
- Jacques, C.E.; Donida, B.; Mescka, C.P.; Rodrigues, D.G.; Marchetti, D.P.; Bitencourt, F.H.; Burin, M.G.; de Souza, C.F.; Giugliani, R.; Vargas, C.R. Oxidative and nitrative stress and pro-inflammatory cytokines in Mucopolysaccharidosis type II patients: Effect of long-term enzyme replacement therapy and relation with glycosaminoglycan accumulation. Biochim. Biophys. Acta 2016, 1862, 1608–1616. [Google Scholar] [CrossRef]
- Raymond, G.V.; Pasquali, M.; Polgreen, L.E.; Dickson, P.I.; Miller, W.P.; Orchard, P.J.; Lund, T.C. Elevated cerebral spinal fluid biomarkers in children with mucopolysaccharidosis I-H. Sci. Rep. 2016, 6, 38305. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Chang, Y.S.; Sung, D.K.; Ko, A.R.; Kim, C.H.; Yoo, D.K.; Lim, K.H.; Sohn, Y.B.; Jin, D.K.; Park, W.S. High-dose enzyme replacement therapy attenuates cerebroventriculomegaly in a mouse model of mucopolysaccharidosis type II. J. Hum. Genet. 2013, 58, 728–733. [Google Scholar] [CrossRef]
- Hamano, K.; Hayashi, M.; Shioda, K.; Fukatsu, R.; Mizutani, S. Mechanisms of neurodegeneration in mucopolysaccharidoses II and IIIB: Analysis of human brain tissue. Acta Neuropathol. 2008, 115, 547–559. [Google Scholar] [CrossRef]
- Corte, A.D.; de Souza, C.F.M.; Anés, M.; Maeda, F.K.; Lokossou, A.; Vedolin, L.M.; Longo, M.G.; Ferreira, M.M.; Perrone, S.G.P.; Balédent, O.; et al. Correlation of CSF flow using phase-contrast MRI with ventriculomegaly and CSF opening pressure in mucopolysaccharidoses. Fluids Barriers CNS 2017, 14, 23. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Zhu, Y.; Hsiao-Nakamoto, J.; Tang, X.; Dugas, J.C.; Moscovitch-Lopatin, M.; Glass, J.D.; Brown, R.H., Jr.; Ladha, S.S.; Lacomis, D.; et al. Longitudinal biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020. [Google Scholar] [CrossRef]
- Bacioglu, M.; Maia, L.F.; Preische, O.; Schelle, J.; Apel, A.; Kaeser, S.A.; Schweighauser, M.; Eninger, T.; Lambert, M.; Pilotto, A.; et al. Neurofilament Light Chain in Blood and CSF as Marker of Disease Progression in Mouse Models and in Neurodegenerative Diseases. Neuron 2016, 91, 56–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Lombardi, V.; Malaspina, A. Neurofilament light: A candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann. Neurol. 2018, 84, 130–139. [Google Scholar] [CrossRef]
- Pawlitzki, M.; Schreiber, S.; Bittner, D.; Kreipe, J.; Leypoldt, F.; Rupprecht, K.; Carare, R.O.; Meuth, S.G.; Vielhaber, S.; Körtvélyessy, P. CSF Neurofilament Light Chain Levels in Primary Progressive MS: Signs of Axonal Neurodegeneration. Front. Neurol. 2018, 9, 1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunnarsson, M.; Malmeström, C.; Axelsson, M.; Sundström, P.; Dahle, C.; Vrethem, M.; Olsson, T.; Piehl, F.; Norgren, N.; Rosengren, L.; et al. Axonal damage in relapsing multiple sclerosis is markedly reduced by natalizumab. Ann. Neurol. 2011, 69, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Alberg, L.; Cullen, N.C.; Michael, E.; Wahlgren, L.; Kroksmark, A.-K.; Rostasy, K.; Blennow, K.; Zetterberg, H.; Tulinius, M. NFL is a marker of treatment response in children with SMA treated with nusinersen. J. Neurol. 2019, 266, 2129–2136. [Google Scholar] [CrossRef] [Green Version]
- Ru, Y.; Corado, C.; Soon, R.K., Jr.; Melton, A.C.; Harris, A.; Yu, G.K.; Pryer, N.; Sinclair, J.R.; Katz, M.L.; Ajayi, T.; et al. Neurofilament light is a treatment-responsive biomarker in CLN2 disease. Ann. Clin. Transl. Neurol. 2019, 6, 2437–2447. [Google Scholar] [CrossRef] [Green Version]
- Scarpa, M.; Orchard, P.J.; Schulz, A.; Dickson, P.I.; Haskins, M.E.; Escolar, M.L.; Giugliani, R. Treatment of brain disease in the mucopolysaccharidoses. Mol. Genet. Metab. 2017, 122s, 25–34. [Google Scholar] [CrossRef]
- Parini, R.; Deodato, F. Intravenous Enzyme Replacement Therapy in Mucopolysaccharidoses: Clinical Effectiveness and Limitations. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Muenzer, J.; Hendriksz, C.J.; Fan, Z.; Vijayaraghavan, S.; Perry, V.; Santra, S.; Solanki, G.A.; Mascelli, M.A.; Pan, L.; Wang, N.; et al. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet. Med. 2016, 18, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Okuyama, T.; Eto, Y.; Sakai, N.; Minami, K.; Yamamoto, T.; Sonoda, H.; Yamaoka, M.; Tachibana, K.; Hirato, T.; Sato, Y. Iduronate-2-Sulfatase with Anti-human Transferrin Receptor Antibody for Neuropathic Mucopolysaccharidosis II: A Phase 1/2 Trial. Mol. Ther. 2019, 27, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Ullman, J.C.; Arguello, A.; Getz, J.A.; Bhalla, A.; Mahon, C.S.; Wang, J.; Giese, T.; Bedard, C.; Kim, D.J.; Blumenfeld, J.R.; et al. Brain delivery and activity of a lysosomal enzyme using a blood-brain barrier transport vehicle in mice. Sci. Transl. Med. 2020, 12, eaay1163. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Lamsa, J.C.; Garcia, A.; Dacosta, J.; Garcia, J.; Treco, D.A. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): A preliminary report. Acta Paediatr. Suppl. 2002, 91, 98–99. [Google Scholar] [CrossRef] [PubMed]
- Kariolis, M.S.; Wells, R.C.; Getz, J.A.; Kwan, W.; Mahon, C.S.; Tong, R.; Kim, D.J.; Srivastava, A.; Bedard, C.; Henne, K.R.; et al. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci. Transl. Med. 2020, 12, eaay1359. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Bigger, B.W. The role of innate immunity in mucopolysaccharide diseases. J. Neurochem. 2019, 148, 639–651. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.; Chen, M.; Zhang, Z.; Corte, A.D.; Souza, C.; Giugliani, R.; Pan, L.; Qiu, Y.; Amaravadi, L.; Wu, J. A novel LC-MS/MS assay to quantify dermatan sulfate in cerebrospinal fluid as a biomarker for mucopolysaccharidosis II. Bioanalysis 2018, 10, 825–838. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Xiao, Y.; Ma, M.; Hu, W.; Liang, T.; Lin, Z.J. Development and validation of an LC-MS/MS Method for the quantitation of heparan sulfate in human urine. Biomed. Chromatogr. 2018, 32, e4294. [Google Scholar] [CrossRef]
- Prasad, V.K.; Kurtzberg, J. Transplant outcomes in mucopolysaccharidoses. Semin. Hematol. 2010, 47, 59–69. [Google Scholar] [CrossRef]
- McKinnis, E.J.; Sulzbacher, S.; Rutledge, J.C.; Sanders, J.; Scott, C.R. Bone marrow transplantation in Hunter syndrome. J. Pediatr. 1996, 129, 145–148. [Google Scholar] [CrossRef]
- Eisengart, J.B.; Rudser, K.D.; Xue, Y.; Orchard, P.; Miller, W.; Lund, T.; Van der Ploeg, A.; Mercer, J.; Jones, S.; Mengel, K.E.; et al. Long-term outcomes of systemic therapies for Hurler syndrome: An international multicenter comparison. Genet. Med. 2018, 20, 1423–1429. [Google Scholar] [CrossRef] [Green Version]
- Kubaski, F.; Yabe, H.; Suzuki, Y.; Seto, T.; Hamazaki, T.; Mason, R.W.; Xie, L.; Onsten, T.G.H.; Leistner-Segal, S.; Giugliani, R.; et al. Hematopoietic Stem Cell Transplantation for Patients with Mucopolysaccharidosis II. Biol. Blood Marrow Transplant. 2017, 23, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.F. Constructing Confidence Sets Using Rank Statistics. J. Am. Stat. Assoc. 1972, 67, 687–690. [Google Scholar] [CrossRef]
- Gallala, H.D.; Sandhoff, K. Biological Function of the Cellular Lipid BMP—BMP as a Key Activator for Cholesterol Sorting and Membrane Digestion. Neurochem. Res. 2011, 36, 1594–1600. [Google Scholar] [CrossRef] [PubMed]
- Daniotti, J.L.; Iglesias-Bartolomé, R. Metabolic pathways and intracellular trafficking of gangliosides. IUBMB Life 2011, 63, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Schulze, H.; Kolter, T.; Sandhoff, K. Principles of lysosomal membrane degradation: Cellular topology and biochemistry of lysosomal lipid degradation. Biochim. Biophys. Acta 2009, 1793, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yu, C.; Sun, Y.; Li, Y. Serum ubiquitin C-terminal hydrolase L1 as a biomarker for traumatic brain injury: A systematic review and meta-analysis. Am. J. Emerg. Med. 2015, 33, 1191–1196. [Google Scholar] [CrossRef]
- Ishiki, A.; Kamada, M.; Kawamura, Y.; Terao, C.; Shimoda, F.; Tomita, N.; Arai, H.; Furukawa, K. Glial fibrillar acidic protein in the cerebrospinal fluid of Alzheimer’s disease, dementia with Lewy bodies, and frontotemporal lobar degeneration. J. Neurochem. 2016, 136, 258–261. [Google Scholar] [CrossRef] [Green Version]
- Heller, C.; Foiani, M.S.; Moore, K.; Convery, R.; Bocchetta, M.; Neason, M.; Cash, D.M.; Thomas, D.; Greaves, C.V.; Woollacott, I.O.; et al. Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2020, 91, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Wuolikainen, A.; Wu, J.; Öhman, A.; Wingsle, G.; Moritz, T.; Andersen, P.M.; Forsgren, L.; Trupp, M. Targeted Multiple Reaction Monitoring Analysis of CSF Identifies UCHL1 and GPNMB as Candidate Biomarkers for ALS. J. Mol. Neurosci. 2019, 69, 643–657. [Google Scholar] [CrossRef] [Green Version]
- Laird, N.M.; Ware, J.H. Random-Effects Models for Longitudinal Data. Biometrics 1982, 38, 963–974. [Google Scholar] [CrossRef]
- Pinheiro, J.C.; Bates, D.; DebRoy, S. The R Core Team nlme: Linear and Nonlinear Mixed Effects Models. R Package Nlme Vers. 2010, 3, 1–97. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CSF | Serum | |||||
|---|---|---|---|---|---|---|
| Baseline Characteristics | Non-MPS Controls | MPS II (ERT) | MPS II (HSCT) a | Non-MPS Controls | MPS II (on ERT) | MPS II (HSCT) a |
| Gender | ||||||
| Male | 25 (100%) | 6 (100%) | 2 (100%) | 7 (47%) | 9 (100%) | 2 (100%) |
| Female | 0 (0%) | 0 (0%) | 0 (0%) | 8 (53%) | 0 (0%) | 0 (0%) |
| Age (years) | ||||||
| Mean (SD) b | 5.8 (5.4) | 3.3 (1.4) | 1 (0) | 5.4 (2.7) | 3.5 (1.2) | 1 (0) |
| Median (min, max) | 5 (<1, 16) | 4 (0.5, 4) | 5 (1, 9) | 4 (0.5, 4) | ||
| N (# of repeated samples) | 25 (0) | 6 (0) | 1 (4) | 15 (0) | 9 (0) | 1 (4) |
| 1 (4) | 1 (6) | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhalla, A.; Ravi, R.; Fang, M.; Arguello, A.; Davis, S.S.; Chiu, C.-L.; Blumenfeld, J.R.; Nguyen, H.N.; Earr, T.K.; Wang, J.; et al. Characterization of Fluid Biomarkers Reveals Lysosome Dysfunction and Neurodegeneration in Neuronopathic MPS II Patients. Int. J. Mol. Sci. 2020, 21, 5188. https://doi.org/10.3390/ijms21155188
Bhalla A, Ravi R, Fang M, Arguello A, Davis SS, Chiu C-L, Blumenfeld JR, Nguyen HN, Earr TK, Wang J, et al. Characterization of Fluid Biomarkers Reveals Lysosome Dysfunction and Neurodegeneration in Neuronopathic MPS II Patients. International Journal of Molecular Sciences. 2020; 21(15):5188. https://doi.org/10.3390/ijms21155188
Chicago/Turabian StyleBhalla, Akhil, Ritesh Ravi, Meng Fang, Annie Arguello, Sonnet S. Davis, Chi-Lu Chiu, Jessica R. Blumenfeld, Hoang N. Nguyen, Timothy K. Earr, Junhua Wang, and et al. 2020. "Characterization of Fluid Biomarkers Reveals Lysosome Dysfunction and Neurodegeneration in Neuronopathic MPS II Patients" International Journal of Molecular Sciences 21, no. 15: 5188. https://doi.org/10.3390/ijms21155188