Mechanisms of Action for Small Molecules Revealed by Structural Biology in Drug Discovery


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
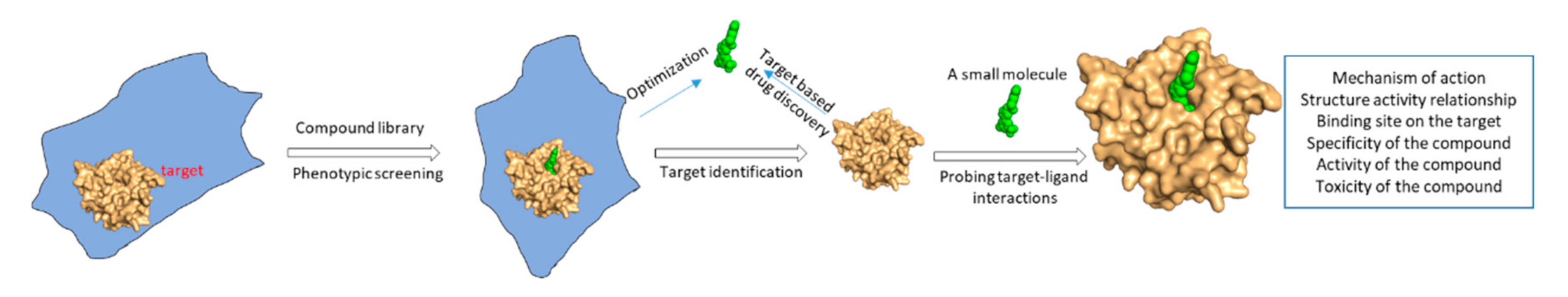
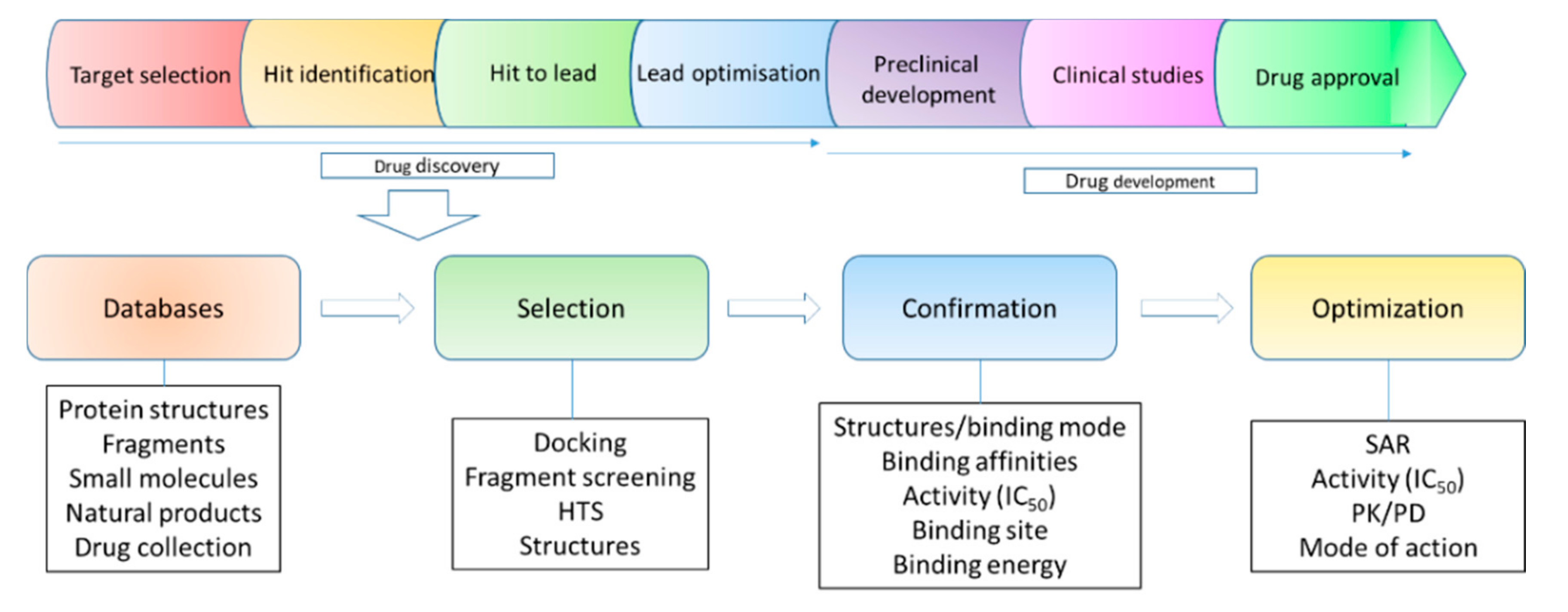
2. Structural Biology in Drug Discovery
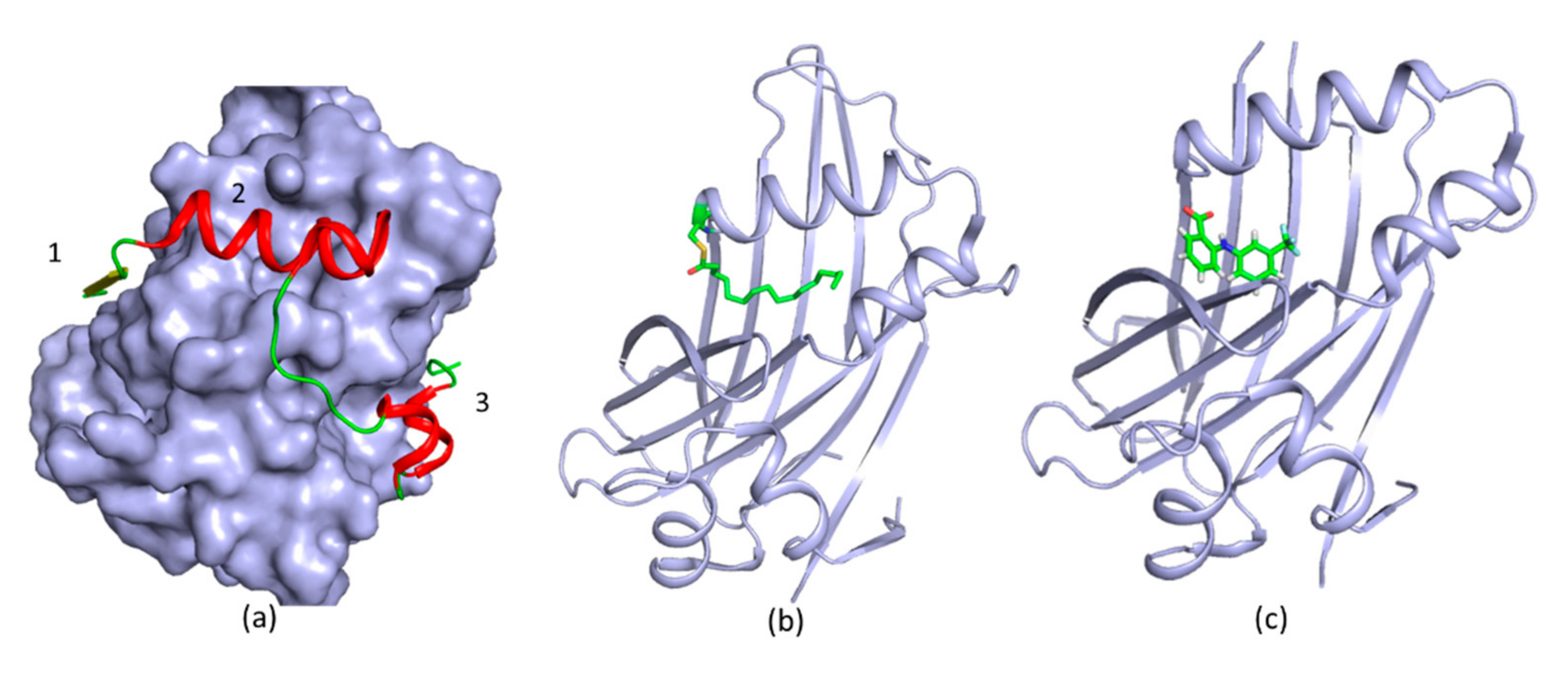
3. New Druggable Sites and Novel Function Derived from Structures
4. Different Types of Protease Inhibitors
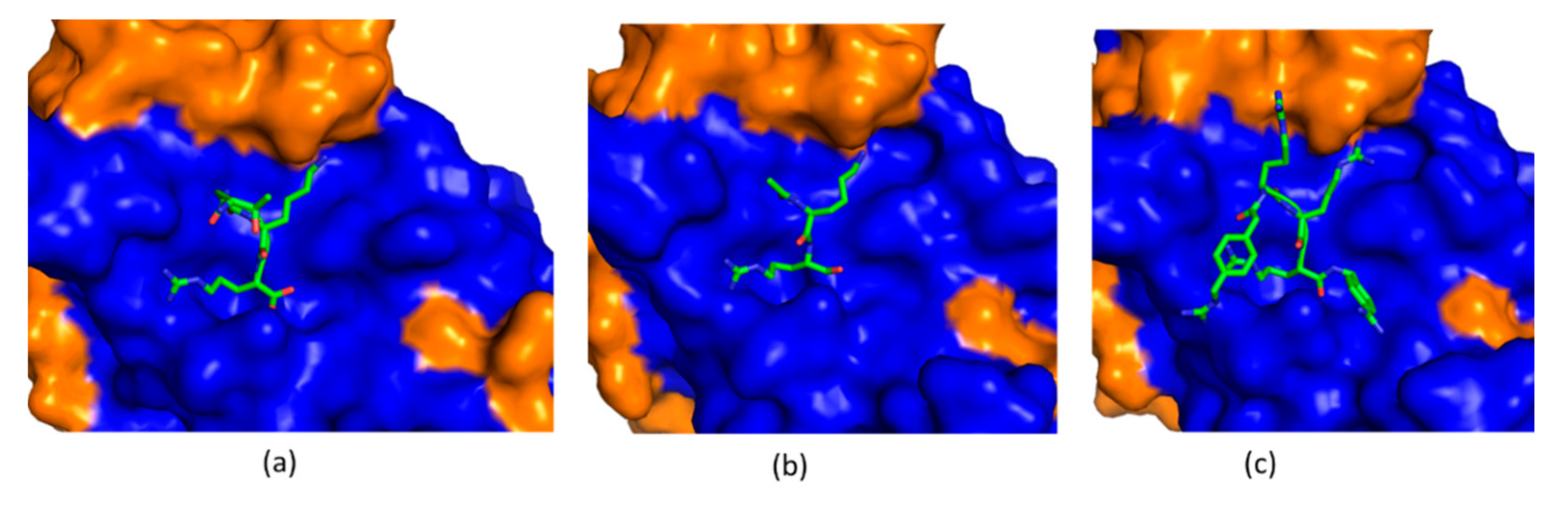
4.1. Substrate-Derived Protease Inhibitors
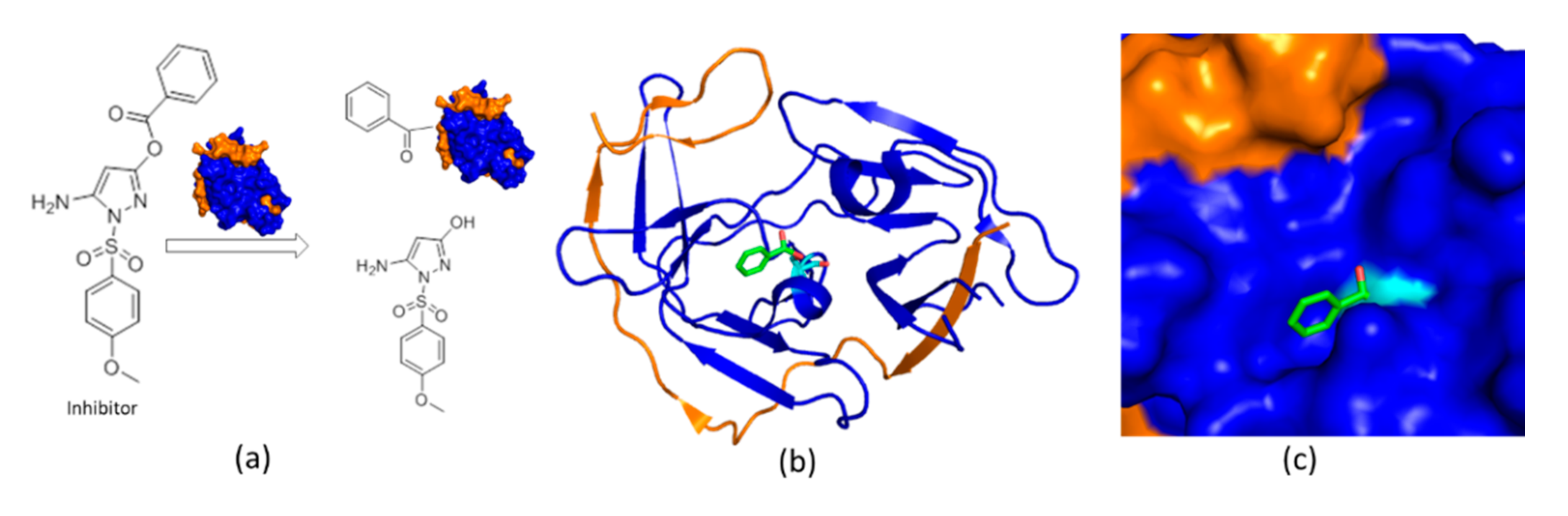
4.2. An Irreversible Small-Molecule Protease Inhibitor
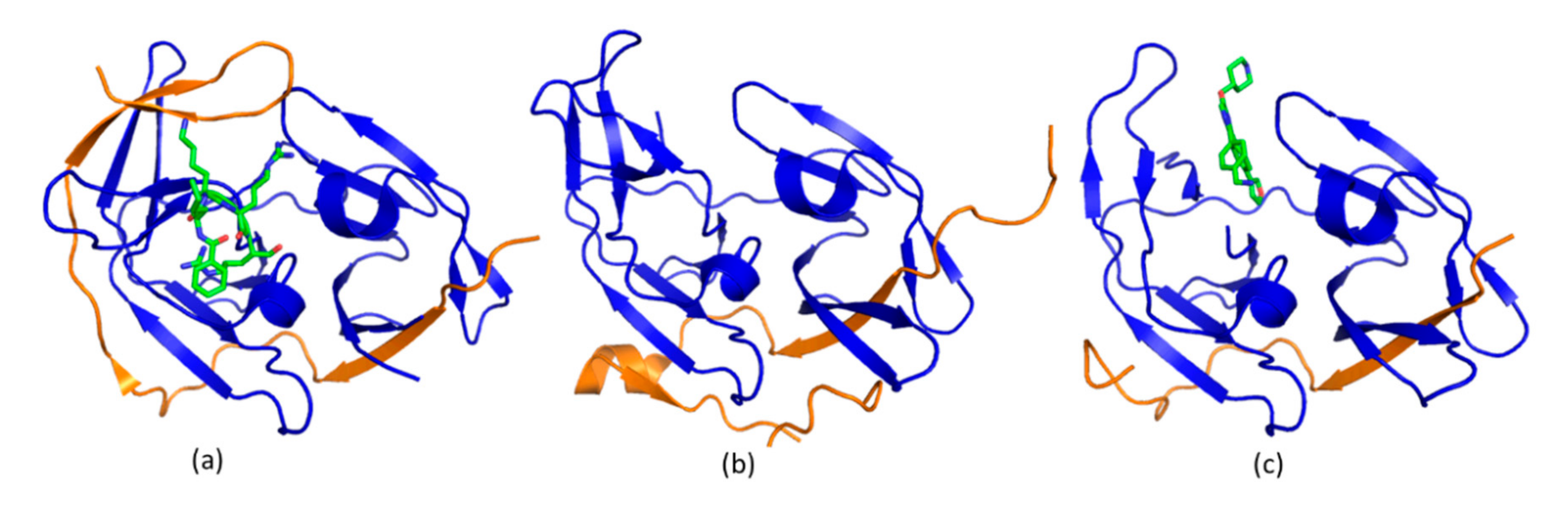
4.3. Allosteric Protease Inhibitors
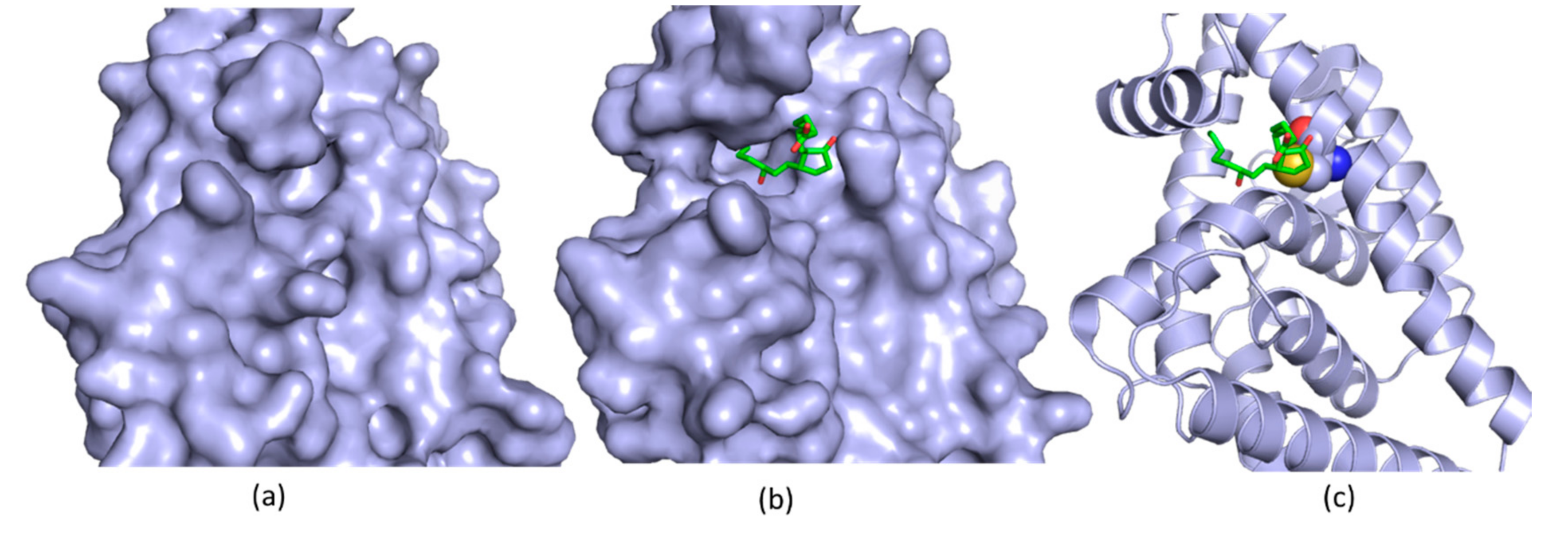
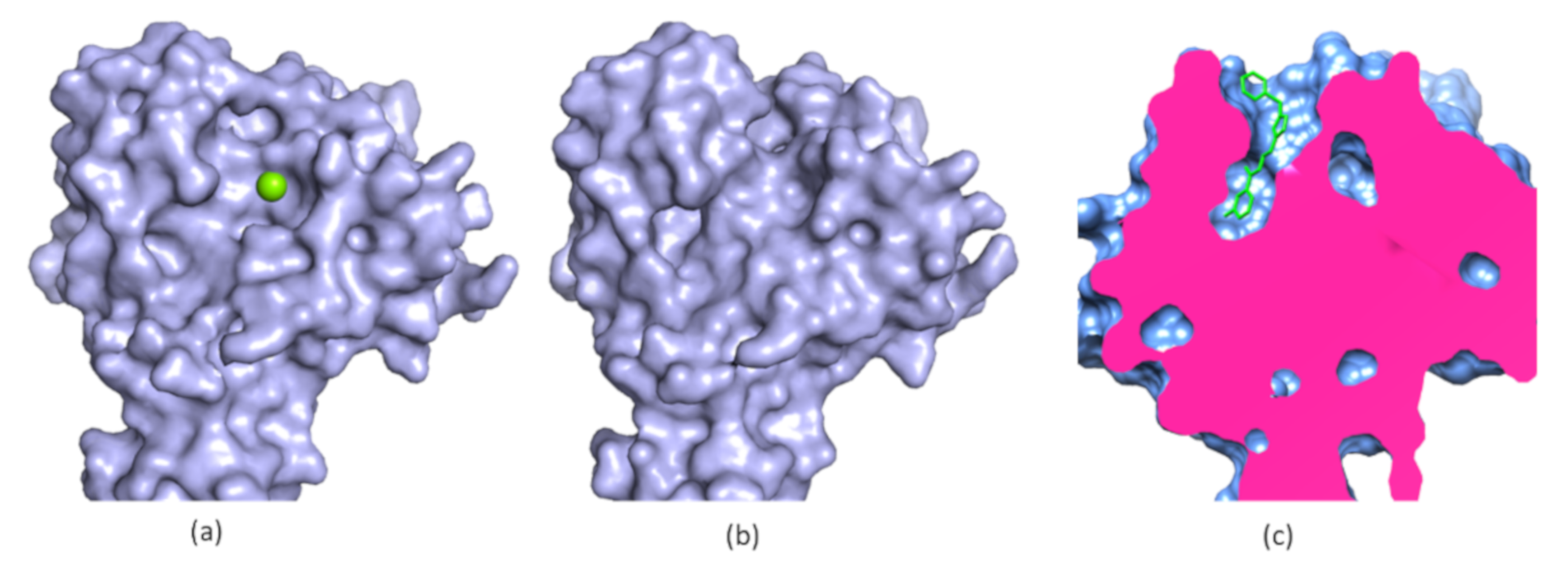
5. Allosteric Inhibitors for a Phosphatase
6. Characterization of On- and Off-Target Effects
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PAINS | Pan-assay interference compounds |
| HTS | High throughput screening |
| NMR | Nuclear magnetic resonance |
| Cryo-EM | Cryogenic electron microscopy |
| SAR | Structure–activity relationship |
| YAP | Yes-associated protein |
| PDB | Protein databank |
| HAD | Haloacid dehalogenase |
| TEAD | Transcriptional enhancer factor with TEA/ATTS domain |
| ZIKV | Zika virus |
| PK | Pharmacokinetic |
| EYA | Eye absent |
| PD | pharmacodynamic |
| Six | Sine oculis homeobox |
References
- Stockwell, B.R. Exploring biology with small organic molecules. Nature 2004, 432, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol. 2012, 6, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Daniels, D.L.; Riching, K.M.; Urh, M. Monitoring and deciphering protein degradation pathways inside cells. Drug Discov. Today Technol. 2019, 31, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein–protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.K.; Schreiber, S.L. The Power of Sophisticated Phenotypic Screening and Modern Mechanism-of-Action Methods. Cell Chem. Biol. 2016, 23, 3–9. [Google Scholar] [CrossRef]
- Wagner, B.K. The resurgence of phenotypic screening in drug discovery and development. Expert Opin. Drug Discov. 2016, 11, 121–125. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Clemons, P.A.; Wagner, B.K. Integrating phenotypic small-molecule profiling and human genetics: The next phase in drug discovery. Trends Genet. 2015, 31, 16–23. [Google Scholar] [CrossRef]
- Jang, B.S. MicroSPECT and MicroPET Imaging of Small Animals for Drug Development. Toxicol. Res. 2013, 29, 1–6. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Brown, D.G.; Boström, J. Where Do Recent Small Molecule Clinical Development Candidates Come from? J. Med. Chem. 2018, 61, 9442–9468. [Google Scholar] [CrossRef] [PubMed]
- Lombardino, J.G.; Lowe, J.A. The role of the medicinal chemist in drug discovery—Then and now. Nat. Rev. Drug Discov. 2004, 3, 853–862. [Google Scholar] [CrossRef] [PubMed]
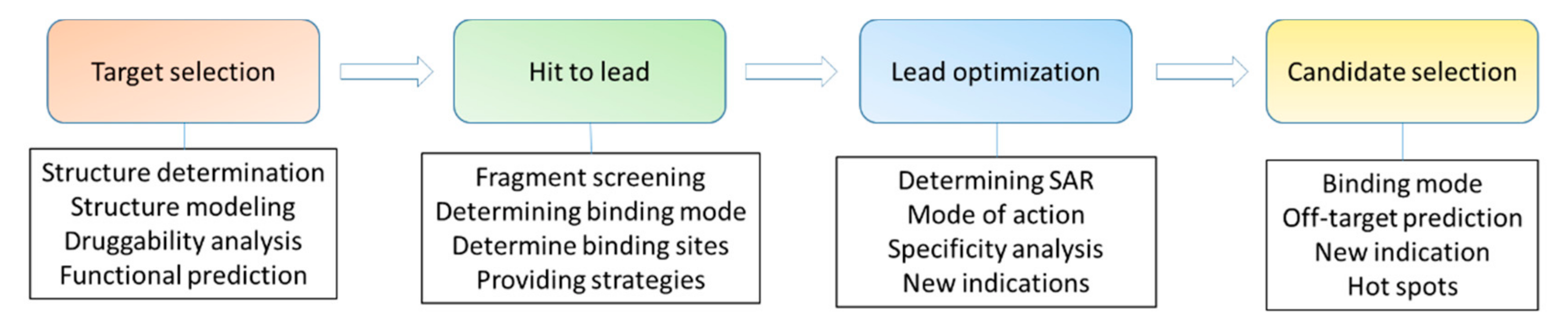
- Stocks, M. Chapter 3—The small molecule drug discovery process—From target selection to candidate selection. In Introduction to Biological and Small Molecule Drug Research and Development; Ganellin, R., Roberts, S., Jefferis, R., Eds.; Elsevier: Oxford, UK, 2013; pp. 81–126. [Google Scholar] [CrossRef]
- Mohs, R.C.; Greig, N.H. Drug discovery and development: Role of basic biological research. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A. High throughput screening in drug discovery. Clin. Transl. Oncol. 2006, 8, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Tam, B.; Akabayov, B. NMR-Fragment Based Virtual Screening: A Brief Overview. Molecules 2018, 23, 233. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef]
- Moitessier, N.; Pottel, J.; Therrien, E.; Englebienne, P.; Liu, Z.; Tomberg, A.; Corbeil, C.R. Medicinal Chemistry Projects Requiring Imaginative Structure-Based Drug Design Methods. Acc. Chem. Res. 2016, 49, 1646–1657. [Google Scholar] [CrossRef]
- Anderson, A.C. The Process of Structure-Based Drug Design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef]
- Ou-Yang, S.-S.; Lu, J.-Y.; Kong, X.-Q.; Liang, Z.-J.; Luo, C.; Jiang, H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. [Google Scholar] [CrossRef]
- Schneider, P.; Walters, W.P.; Plowright, A.T.; Sieroka, N.; Listgarten, J.; Goodnow, R.A.; Fisher, J.; Jansen, J.M.; Duca, J.S.; Rush, T.S.; et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 2020, 19, 353–364. [Google Scholar] [CrossRef]
- Mullard, A. A snapshot of lead-generation strategies. Nat. Rev. Drug Discov. 2018, 17, 534. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Goodnow, R.A.; Dumelin, C.E.; Keefe, A.D. DNA-encoded chemistry: Enabling the deeper sampling of chemical space. Nat. Rev. Drug Discov. 2017, 16, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.I.; McGregor, L.M.; Liu, D.R. Novel selection methods for DNA-encoded chemical libraries. Curr. Opin. Chem. Biol. 2015, 26, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Gerry, C.J.; Wawer, M.J.; Clemons, P.A.; Schreiber, S.L. DNA Barcoding a Complete Matrix of Stereoisomeric Small Molecules. J. Am. Chem. Soc. 2019, 141, 10225–10235. [Google Scholar] [CrossRef]
- Dahlin, J.L.; Nissink, J.W.M.; Strasser, J.M.; Francis, S.; Higgins, L.; Zhou, H.; Zhang, Z.; Walters, M.A. PAINS in the Assay: Chemical Mechanisms of Assay Interference and Promiscuous Enzymatic Inhibition Observed during a Sulfhydryl-Scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. [Google Scholar] [CrossRef]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef]
- Hughes, J.; Rees, S.; Kalindjian, S.; Philpott, K. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- An, W.F.; Tolliday, N. Cell-Based Assays for High-Throughput Screening. Mol. Biotechnol. 2010, 45, 180–186. [Google Scholar] [CrossRef]
- Duggirala, N.K.; Perry, M.L.; Almarsson, Ö.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medicines. Chem. Commun. 2016, 52, 640–655. [Google Scholar] [CrossRef]
- Congreve, M.; Murray, C.W.; Blundell, T.L. Keynote review: Structural biology and drug discovery. Drug Discov. Today 2005, 10, 895–907. [Google Scholar] [CrossRef]
- Evanthia, L.; George, S.; Demetrios, K.V.; Zoe, C. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Brunger, A.T. X-ray crystallography and NMR reveal complementary views of structure and dynamics. Nat. Struct. Biol. 1997, 4, 862–865. [Google Scholar]
- Cassiday, L. Structural biology: More than a crystallographer. Nature 2014, 505, 711–713. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. A Glimpse of Structural Biology through X-Ray Crystallography. Cell 2014, 159, 995–1014. [Google Scholar] [CrossRef]
- Wagner, G. An account of NMR in structural biology. Nat. Struct. Biol. 1997, 4, 841–844. [Google Scholar] [PubMed]
- Howard, M.J. Protein NMR spectroscopy. Curr. Biol. 1998, 8, R331–R333. [Google Scholar] [CrossRef]
- Nogales, E. The development of cryo-EM into a mainstream structural biology technique. Nat. Methods 2016, 13, 24–27. [Google Scholar] [CrossRef]
- Callaway, E. The revolution will not be crystallized: A new method sweeps through structural biology. Nature 2015, 525, 172–174. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.A.; Strauss, C.E.M.; Misura, K.M.S.; Baker, D. Protein Structure Prediction Using Rosetta. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2004; Volume 383, pp. 66–93. [Google Scholar]
- Zheng, H.; Hou, J.; Zimmerman, M.D.; Wlodawer, A.; Minor, W. The future of crystallography in drug discovery. Expert Opin. Drug Discov. 2014, 9, 125–137. [Google Scholar] [CrossRef]
- Pellecchia, M.; Sem, D.S.; Wuthrich, K. NMR in drug discovery. Nat. Rev. Drug Discov. 2002, 1, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Fesik, S.W.; Zuiderweg, E.R.; Olejniczak, E.T.; Gampe, R.T., Jr. NMR methods for determining the structures of enzyme/inhibitor complexes as an aid in drug design. Biochem. Pharmacol. 1990, 40, 161–167. [Google Scholar] [CrossRef]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef]
- Harner, M.J.; Frank, A.O.; Fesik, S.W. Fragment-based drug discovery using NMR spectroscopy. J. Biomol. NMR 2013, 56, 65–75. [Google Scholar] [CrossRef]
- Matthies, D.; Bae, C.; Toombes, G.E.S.; Fox, T.; Bartesaghi, A.; Subramaniam, S.; Swartz, K.J. Single-particle cryo-EM structure of a voltage-activated potassium channel in lipid nanodiscs. eLife 2018, 7, e37558. [Google Scholar] [CrossRef]
- Ceska, T.; Chung, C.-W.; Cooke, R.; Phillips, C.; Williams, P.A. Cryo-EM in drug discovery. Biochem. Soc. Trans. 2019, 47, 281–293. [Google Scholar] [CrossRef]
- Fauman, E.B.; Rai, B.K.; Huang, E.S. Structure-based druggability assessment—Identifying suitable targets for small molecule therapeutics. Curr. Opin. Chem. Biol. 2011, 15, 463–468. [Google Scholar] [CrossRef]
- Unver, M.Y.; Gierse, R.M.; Ritchie, H.; Hirsch, A.K.H. Druggability Assessment of Targets Used in Kinetic Target-Guided Synthesis. J. Med. Chem. 2018, 61, 9395–9409. [Google Scholar] [CrossRef]
- Thomas, S.E.; Collins, P.; James, R.H.; Mendes, V.; Charoensutthivarakul, S.; Radoux, C.; Abell, C.; Coyne, A.G.; Floto, R.A.; Delft, F.v.; et al. Structure-guided fragment-based drug discovery at the synchrotron: Screening binding sites and correlations with hotspot mapping. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2019, 377, 20180422. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, C.; Stahl, E.; Koessel, K.; Rivera, A.; Cherry, B.R.; Pulavarti, S.V.S.R.K.; Szyperski, T.; Cance, W.; Marlowe, T. Development of a Fragment-Based Screening Assay for the Focal Adhesion Targeting Domain Using SPR and NMR. Molecules 2019, 24, 3352. [Google Scholar] [CrossRef]
- Nitsche, C.; Otting, G. NMR studies of ligand binding. Curr. Opin. Struct. Biol. 2018, 48, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S.; Leung, E.W.; Chandrashekaran, I.R.; MacRaild, C.A. Applications of (19)F-NMR in Fragment-Based Drug Discovery. Molecules 2016, 21, 860. [Google Scholar] [CrossRef] [PubMed]
- McKinney, J.D.; Richard, A.; Waller, C.; Newman, M.C.; Gerberick, F. The Practice of Structure Activity Relationships (SAR) in Toxicology. Toxicol. Sci. 2000, 56, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M. High-throughput docking for lead generation. Curr. Opin. Chem. Biol. 2001, 5, 375–382. [Google Scholar] [CrossRef]
- Macalino, S.J.Y.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Wang, X.; Song, K.; Li, L.; Chen, L. Structure-Based Drug Design Strategies and Challenges. Curr. Top. Med. Chem. 2018, 18, 998–1006. [Google Scholar] [CrossRef]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [PubMed]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Fu, V.; Plouffe, S.W.; Guan, K.L. The Hippo pathway in organ development, homeostasis, and regeneration. Curr. Opin. Cell Biol. 2018, 49, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Guan, K.L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Santucci, M.; Vignudelli, T.; Ferrari, S.; Mor, M.; Scalvini, L.; Bolognesi, M.L.; Uliassi, E.; Costi, M.P. The Hippo Pathway and YAP/TAZ–TEAD Protein–Protein Interaction as Targets for Regenerative Medicine and Cancer Treatment. J. Med. Chem. 2015, 58, 4857–4873. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Yin, M.; Zhang, L. Hippo signaling: A hub of growth control, tumor suppression and pluripotency maintenance. J. Genet. Genom. 2011, 38, 471–481. [Google Scholar] [CrossRef]
- Cairns, L.; Tran, T.; Kavran, J.M. Structural Insights into the Regulation of Hippo Signaling. ACS Chem. Biol. 2017, 12, 601–610. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, B.; Wang, P.; Chen, F.; Dong, Z.; Yang, H.; Guan, K.L.; Xu, Y. Structural insights into the YAP and TEAD complex. Genes Dev. 2010, 24, 235–240. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Chan, S.W.; Lee, I.; Song, H.; Hong, W. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure 2012, 20, 1135–1140. [Google Scholar] [CrossRef]
- Kaan, H.Y.K.; Chan, S.W.; Tan, S.K.J.; Guo, F.; Lim, C.J.; Hong, W.; Song, H. Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Sci. Rep. 2017, 7, 2035. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Han, X.; Hung, A.W.; Weiguang, S.; Huda, N.; Chen, G.Y.; Kang, C.; Chia, C.S.; Luo, X.; Hong, W.; et al. Targeting the Central Pocket in Human Transcription Factor TEAD as a Potential Cancer Therapeutic Strategy. Structure 2015, 23, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hu, T.; Xu, Z.; Lin, Z.; Zhang, Z.; Feng, T.; Zhu, L.; Rong, Y.; Shen, H.; Luk, J.M.; et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lin, Z.; Zhou, Z.; Shen, H.C.; Yan, S.F.; Mayweg, A.V.; Xu, Z.; Qin, N.; Wong, J.C.; Zhang, Z.; et al. Structure-Based Design and Synthesis of Potent Cyclic Peptides Inhibiting the YAP–TEAD Protein–Protein Interaction. ACS Med. Chem. Lett. 2014, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A Peptide Mimicking VGLL4 Function Acts as a YAP Antagonist Therapy against Gastric Cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Crook, Z.R.; Sevilla, G.P.; Friend, D.; Brusniak, M.Y.; Bandaranayake, A.D.; Clarke, M.; Gewe, M.; Mhyre, A.J.; Baker, D.; Strong, R.K.; et al. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat. Commun. 2017, 8, 2244. [Google Scholar] [CrossRef]
- Noland, C.L.; Gierke, S.; Schnier, P.D.; Murray, J.; Sandoval, W.N.; Sagolla, M.; Dey, A.; Hannoush, R.N.; Fairbrother, W.J.; Cunningham, C.N. Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure 2016, 24, 179–186. [Google Scholar] [CrossRef]
- Gibault, F.; Sturbaut, M.; Bailly, F.; Melnyk, P.; Cotelle, P. Targeting Transcriptional Enhanced Associate Domains (TEADs). J. Med. Chem. 2018, 61, 5057–5072. [Google Scholar] [CrossRef]
- Kunig, V.B.K.; Potowski, M.; Akbarzadeh, M.; Klika Škopić, M.; Dos Santos Smith, D.; Arendt, L.; Dormuth, I.; Adihou, H.; Andlovic, B.; Karatas, H.; et al. TEAD-YAP interaction inhibitors and MDM2 binders from DNA-encoded indole-focused Ugi-peptidomimetics. Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef]
- Chan, P.; Han, X.; Zheng, B.; DeRan, M.; Yu, J.; Jarugumilli, G.K.; Deng, H.; Pan, D.; Luo, X.; Wu, X. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol. 2016, 12, 282. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.K.; Crawford, J.J.; Noland, C.L.; Schmidt, S.; Zbieg, J.R.; Lacap, J.A.; Zang, R.; Miller, G.M.; Zhang, Y.; Beroza, P.; et al. Small Molecule Dysregulation of TEAD Lipidation Induces a Dominant-Negative Inhibition of Hippo Pathway Signaling. Cell Rep. 2020, 31, 107809. [Google Scholar] [CrossRef] [PubMed]
- Rajan, S.; Jang, Y.; Kim, C.-H.; Kim, W.; Toh, H.T.; Jeon, J.; Song, B.; Serra, A.; Lescar, J.; Yoo, J.Y.; et al. PGE1 and PGA1 bind to Nurr1 and activate its transcriptional function. Nat. Chem. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Anantharajan, J.; Zhou, H.; Zhang, L.; Hotz, T.; Vincent, M.Y.; Blevins, M.A.; Jansson, A.E.; Kuan, J.W.L.; Ng, E.Y.; Yeo, Y.K.; et al. Structural and Functional Analyses of an Allosteric EYA2 Phosphatase Inhibitor That Has On-Target Effects in Human Lung Cancer Cells. Mol. Cancer Ther. 2019, 18, 1484–1496. [Google Scholar] [CrossRef] [PubMed]
- Pearen, M.A.; Muscat, G.E.O. Minireview: Nuclear Hormone Receptor 4A Signaling: Implications for Metabolic Disease. Mol. Endocrinol. 2010, 24, 1891–1903. [Google Scholar] [CrossRef] [PubMed]
- Decressac, M.; Volakakis, N.; Björklund, A.; Perlmann, T. NURR1 in Parkinson disease—From pathogenesis to therapeutic potential. Nat. Rev. Neurol. 2013, 9, 629–636. [Google Scholar] [CrossRef]
- Wang, Z.; Benoit, G.; Liu, J.; Prasad, S.; Aarnisalo, P.; Liu, X.; Xu, H.; Walker, N.P.C.; Perlmann, T. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature 2003, 423, 555–560. [Google Scholar] [CrossRef]
- De Vera, I.M.S.; Munoz-Tello, P.; Zheng, J.; Dharmarajan, V.; Marciano, D.P.; Matta-Camacho, E.; Giri, P.K.; Shang, J.; Hughes, T.S.; Rance, M.; et al. Defining a Canonical Ligand-Binding Pocket in the Orphan Nuclear Receptor Nurr1. Structure 2019, 27, 66–77. [Google Scholar] [CrossRef]
- Windshügel, B. Structural insights into ligand-binding pocket formation in Nurr1 by molecular dynamics simulations. J. Biomol. Struct. Dyn. 2019, 37, 4651–4657. [Google Scholar] [CrossRef]
- Poulsen, A.; Kang, C.; Keller, T.H. Drug design for flavivirus proteases: What are we missing? Curr. Pharm. Des. 2014, 20, 3422–3427. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. Flaviviruses. In Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 747–794. [Google Scholar]
- Lindenbach, B.D.; Thiel, H.J.; Rice, C.M. Flaviviridae. In Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 712–746. [Google Scholar]
- Weaver, S.C.; Costa, F.; Garcia-Blanco, M.A.; Ko, A.I.; Ribeiro, G.S.; Saade, G.; Shi, P.Y.; Vasilakis, N. Zika virus: History, emergence, biology, and prospects for control. Antivir. Res. 2016, 130, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Jamieson, D.J.; Powers, A.M.; Honein, M.A. Zika Virus. N. Engl. J. Med. 2016, 374, 1552–1563. [Google Scholar] [CrossRef] [PubMed]
- Broutet, N.; Krauer, F.; Riesen, M.; Khalakdina, A.; Almiron, M.; Aldighieri, S.; Espinal, M.; Low, N.; Dye, C. Zika Virus as a Cause of Neurologic Disorders. N. Engl. J. Med. 2016, 374, 1506–1509. [Google Scholar] [CrossRef] [PubMed]
- Carteaux, G.; Maquart, M.; Bedet, A.; Contou, D.; Brugieres, P.; Fourati, S.; Cleret de Langavant, L.; de Broucker, T.; Brun-Buisson, C.; Leparc-Goffart, I.; et al. Zika Virus Associated with Meningoencephalitis. N. Engl. J. Med. 2016, 374, 1595–1596. [Google Scholar] [CrossRef]
- Mecharles, S.; Herrmann, C.; Poullain, P.; Tran, T.H.; Deschamps, N.; Mathon, G.; Landais, A.; Breurec, S.; Lannuzel, A. Acute myelitis due to Zika virus infection. Lancet 2016, 387, 1481. [Google Scholar] [CrossRef]
- Baronti, C.; Piorkowski, G.; Charrel, R.N.; Boubis, L.; Leparc-Goffart, I.; de Lamballerie, X. Complete coding sequence of zika virus from a French polynesia outbreak in 2013. Genome Announc. 2014, 2, e00500-14. [Google Scholar] [CrossRef]
- Clum, S.; Ebner, K.E.; Padmanabhan, R. Cotranslational membrane insertion of the serine proteinase precursor NS2B-NS3(Pro) of dengue virus type 2 is required for efficient in vitro processing and is mediated through the hydrophobic regions of NS2B. J. Biol. Chem. 1997, 272, 30715–30723. [Google Scholar] [CrossRef]
- Li, Y.; Li, Q.; Wong, Y.L.; Liew, L.S.; Kang, C. Membrane topology of NS2B of dengue virus revealed by NMR spectroscopy. Biochim. Biophys. Acta 2015, 1848, 2244–2252. [Google Scholar] [CrossRef]
- Brecher, M.; Zhang, J.; Li, H. The flavivirus protease as a target for drug discovery. Virol. Sin. 2013, 28, 326–336. [Google Scholar] [CrossRef]
- Phoo, W.W.; Zhang, Z.; Wirawan, M.; Chew, E.J.C.; Chew, A.B.L.; Kouretova, J.; Steinmetzer, T.; Luo, D. Structures of Zika virus NS2B-NS3 protease in complex with peptidomimetic inhibitors. Antivir. Res. 2018, 160, 17–24. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Farhy, C.; Pinto, A.; Huang, C.-T.; Simonetti, N.; Ngono, A.E.; Dewing, A.; Shresta, S.; Pinkerton, A.B.; Cieplak, P.; et al. Characterization of the Zika virus two-component NS2B-NS3 protease and structure-assisted identification of allosteric small-molecule antagonists. Antivir. Res. 2017, 143, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Y.; Loh, Y.R.; Phoo, W.W.; Hung, A.W.; Kang, C.; Luo, D. Crystal structure of unlinked NS2B-NS3 protease from Zika virus. Science 2016, 354, 1597–1600. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Ji, X.; Yang, X.; Xie, W.; Yang, K.; Chen, C.; Wu, C.; Chi, H.; Mu, Z.; Wang, Z.; et al. The crystal structure of Zika virus helicase: Basis for antiviral drug design. Protein Cell 2016. [Google Scholar] [CrossRef] [PubMed]
- Phoo, W.W.; Li, Y.; Zhang, Z.; Lee, M.Y.; Loh, Y.R.; Tan, Y.B.; Ng, E.Y.; Lescar, J.; Kang, C.; Luo, D. Structure of the NS2B-NS3 protease from Zika virus after self-cleavage. Nat. Commun. 2016, 7, 13410. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Hansen, G.; Nitsche, C.; Klein, C.D.; Zhang, L.; Hilgenfeld, R. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 2016, 353, 503–505. [Google Scholar] [CrossRef]
- Nitsche, C.; Onagi, H.; Quek, J.P.; Otting, G.; Luo, D.; Huber, T. Biocompatible Macrocyclization between Cysteine and 2-Cyanopyridine Generates Stable Peptide Inhibitors. Org. Lett. 2019, 21, 4709–4712. [Google Scholar] [CrossRef] [PubMed]
- Aleshin, A.E.; Shiryaev, S.A.; Strongin, A.Y.; Liddington, R.C. Structural evidence for regulation and specificity of flaviviral proteases and evolution of the Flaviviridae fold. Protein. Sci. 2007, 16, 795–806. [Google Scholar] [CrossRef]
- Chandramouli, S.; Joseph, J.S.; Daudenarde, S.; Gatchalian, J.; Cornillez-Ty, C.; Kuhn, P. Serotype-specific structural differences in the protease-cofactor complexes of the dengue virus family. J. Virol. 2010, 84, 3059–3067. [Google Scholar] [CrossRef]
- Robin, G.; Chappell, K.; Stoermer, M.J.; Hu, S.H.; Young, P.R.; Fairlie, D.P.; Martin, J.L. Structure of West Nile virus NS3 protease: Ligand stabilization of the catalytic conformation. J. Mol. Biol. 2009, 385, 1568–1577. [Google Scholar] [CrossRef]
- Hammamy, M.Z.; Haase, C.; Hammami, M.; Hilgenfeld, R.; Steinmetzer, T. Development and characterization of new peptidomimetic inhibitors of the West Nile virus NS2B-NS3 protease. Chem. Med. Chem. 2013, 8, 231–241. [Google Scholar] [CrossRef]
- Gruba, N.; Martinez, J.I.R.; Grzywa, R.; Wysocka, M.; Skorenski, M.; Dabrowska, A.; Lecka, M.; Suder, P.; Sienczyk, M.; Pyrc, K.; et al. One Step Beyond: Design of Substrates Spanning Primed Positions of Zika Virus NS2B-NS3 Protease. ACS Med. Chem. Lett. 2018, 9, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Gayen, S.; Wang, W.; Severin, R.; Chen, A.S.; Lim, H.A.; Chia, C.S.; Schuller, A.; Doan, D.N.; Poulsen, A.; et al. Exploring the binding of peptidic West Nile virus NS2B-NS3 protease inhibitors by NMR. Antivir. Res. 2013, 97, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kang, C. Insights into Structures and Dynamics of Flavivirus Proteases from NMR Studies. Int. J. Mol. Sci. 2020, 21, 2527. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Z.; Phoo, W.W.; Loh, Y.R.; Wang, W.; Liu, S.; Chen, M.W.; Hung, A.W.; Keller, T.H.; Luo, D.; et al. Structural Dynamics of Zika Virus NS2B-NS3 Protease Binding to Dipeptide Inhibitors. Structure 2017, 25, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Quek, J.P.; Liu, S.; Zhang, Z.; Li, Y.; Ng, E.Y.; Loh, Y.R.; Hung, A.W.; Luo, D.; Kang, C. Identification and structural characterization of small molecule fragments targeting Zika virus NS2B-NS3 protease. Antivir. Res. 2020, 175, 104707. [Google Scholar] [CrossRef]
- Lim, S.P.; Shi, P.Y. West Nile virus drug discovery. Viruses 2013, 5, 2977–3006. [Google Scholar] [CrossRef]
- Lim, S.P.; Wang, Q.Y.; Noble, C.G.; Chen, Y.L.; Dong, H.; Zou, B.; Yokokawa, F.; Nilar, S.; Smith, P.; Beer, D.; et al. Ten years of dengue drug discovery: Progress and prospects. Antivir. Res. 2013, 100, 500–519. [Google Scholar] [CrossRef]
- Nitsche, C.; Behnam, M.A.; Steuer, C.; Klein, C.D. Retro peptide-hybrids as selective inhibitors of the Dengue virus NS2B-NS3 protease. Antivir. Res. 2012, 94, 72–79. [Google Scholar] [CrossRef]
- Yin, Z.; Patel, S.J.; Wang, W.L.; Wang, G.; Chan, W.L.; Rao, K.R.; Alam, J.; Jeyaraj, D.A.; Ngew, X.; Patel, V.; et al. Peptide inhibitors of Dengue virus NS3 protease. Part 1: Warhead. Bioorg. Med. Chem. Lett. 2006, 16, 36–39. [Google Scholar] [CrossRef]
- Deng, J.; Li, N.; Liu, H.; Zuo, Z.; Liew, O.W.; Xu, W.; Chen, G.; Tong, X.; Tang, W.; Zhu, J.; et al. Discovery of novel small molecule inhibitors of dengue viral NS2B-NS3 protease using virtual screening and scaffold hopping. J. Med. Chem. 2012, 55, 6278–6293. [Google Scholar] [CrossRef]
- Yang, C.C.; Hsieh, Y.C.; Lee, S.J.; Wu, S.H.; Liao, C.L.; Tsao, C.H.; Chao, Y.S.; Chern, J.H.; Wu, C.P.; Yueh, A. Novel dengue virus-specific NS2B/NS3 protease inhibitor, BP2109, discovered by a high-throughput screening assay. Antimicrob. Agents Chemother. 2011, 55, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.A.; Phillips, J.; Shun, T.Y.; Shinde, S.; Lazo, J.S.; Huryn, D.M.; Myers, M.C.; Ratnikov, B.I.; Smith, J.W.; Su, Y.; et al. HTS identifies novel and specific uncompetitive inhibitors of the two-component NS2B-NS3 proteinase of West Nile virus. Assay Drug Dev. Technol. 2007, 5, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Sidique, S.; Shiryaev, S.A.; Ratnikov, B.I.; Herath, A.; Su, Y.; Strongin, A.Y.; Cosford, N.D. Structure-activity relationship and improved hydrolytic stability of pyrazole derivatives that are allosteric inhibitors of West Nile Virus NS2B-NS3 proteinase. Bioorg. Med. Chem. Lett. 2009, 19, 5773–5777. [Google Scholar] [CrossRef] [PubMed]
- Koh-Stenta, X.; Joy, J.; Wang, S.F.; Kwek, P.Z.; Wee, J.L.; Wan, K.F.; Gayen, S.; Chen, A.S.; Kang, C.; Lee, M.A.; et al. Identification of covalent active site inhibitors of dengue virus protease. Drug Des. Devel. Ther. 2015, 9, 6389–6399. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Phoo, W.W.; Loh, Y.R.; Li, R.; Yang, H.Y.; Jansson, A.E.; Hill, J.; Keller, T.H.; Nacro, K.; et al. Structural Insights into the Inhibition of Zika Virus NS2B-NS3 Protease by a Small-Molecule Inhibitor. Structure 2018, 26, 555–564. [Google Scholar] [CrossRef]
- Schöne, T.; Grimm, L.L.; Sakai, N.; Zhang, L.; Hilgenfeld, R.; Peters, T. STD-NMR experiments identify a structural motif with novel second-site activity against West Nile virus NS2B-NS3 protease. Antivir. Res. 2017, 146, 174–183. [Google Scholar] [CrossRef]
- Kim, Y.M.; Gayen, S.; Kang, C.; Joy, J.; Huang, Q.; Chen, A.S.; Wee, J.L.; Ang, M.J.; Lim, H.A.; Hung, A.W.; et al. NMR analysis of a novel enzymatically active unlinked dengue NS2B-NS3 protease complex. J. Biol. Chem. 2013, 288, 12891–12900. [Google Scholar] [CrossRef]
- Su, X.C.; Ozawa, K.; Qi, R.; Vasudevan, S.G.; Lim, S.P.; Otting, G. NMR analysis of the dynamic exchange of the NS2B cofactor between open and closed conformations of the West Nile virus NS2B-NS3 protease. PLoS Negl. Trop. Dis. 2009, 3, e561. [Google Scholar] [CrossRef]
- Nitsche, C.; Passioura, T.; Varava, P.; Mahawaththa, M.C.; Leuthold, M.M.; Klein, C.D.; Suga, H.; Otting, G. De Novo Discovery of Nonstandard Macrocyclic Peptides as Noncompetitive Inhibitors of the Zika Virus NS2B-NS3 Protease. ACS Med. Chem. Lett. 2019, 10, 168–174. [Google Scholar] [CrossRef]
- Yao, Y.; Huo, T.; Lin, Y.-L.; Nie, S.; Wu, F.; Hua, Y.; Wu, J.; Kneubehl, A.R.; Vogt, M.B.; Rico-Hesse, R.; et al. Discovery, X-ray Crystallography and Antiviral Activity of Allosteric Inhibitors of Flavivirus NS2B-NS3 Protease. J. Am. Chem. Soc. 2019, 141, 6832–6836. [Google Scholar] [CrossRef]
- Yildiz, M.; Ghosh, S.; Bell, J.A.; Sherman, W.; Hardy, J.A. Allosteric Inhibition of the NS2B-NS3 Protease from Dengue Virus. ACS Chem. Biol. 2013, 8, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Othman, R.; Kiat, T.S.; Khalid, N.; Yusof, R.; Newhouse, E.I.; Newhouse, J.S.; Alam, M.; Rahman, N.A. Docking of noncompetitive inhibitors into dengue virus type 2 protease: Understanding the interactions with allosteric binding sites. J. Chem. Inf. Model 2008, 48, 1582–1591. [Google Scholar] [CrossRef] [PubMed]
- Ekonomiuk, D.; Su, X.C.; Ozawa, K.; Bodenreider, C.; Lim, S.P.; Otting, G.; Huang, D.; Caflisch, A. Flaviviral protease inhibitors identified by fragment-based library docking into a structure generated by molecular dynamics. J. Med. Chem. 2009, 52, 4860–4868. [Google Scholar] [CrossRef] [PubMed]
- Ekonomiuk, D.; Su, X.C.; Ozawa, K.; Bodenreider, C.; Lim, S.P.; Yin, Z.; Keller, T.H.; Beer, D.; Patel, V.; Otting, G.; et al. Discovery of a non-peptidic inhibitor of west nile virus NS3 protease by high-throughput docking. PLoS Negl. Trop. Dis. 2009, 3, e356. [Google Scholar] [CrossRef]
- Knox, J.E.; Ma, N.L.; Yin, Z.; Patel, S.J.; Wang, W.L.; Chan, W.L.; Rao, K.R.R.; Wang, G.; Ngew, X.; Patel, V.; et al. Peptide inhibitors of West Nile NS3 protease: SAR study of tetrapeptide aldehyde inhibitors. J. Med. Chem. 2006, 49, 6585–6590. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, L.; Vartuli, R.L.; Ford, H.L.; Zhao, R. The Eya phosphatase: Its unique role in cancer. Int. J. Biochem. Cell Biol. 2018, 96, 165–170. [Google Scholar] [CrossRef]
- Patrick, A.N.; Cabrera, J.H.; Smith, A.L.; Chen, X.S.; Ford, H.L.; Zhao, R. Structure-function analyses of the human SIX1–EYA2 complex reveal insights into metastasis and BOR syndrome. Nat. Struct. Mol. Biol. 2013, 20, 447–453. [Google Scholar] [CrossRef]
- Kumar, J.P. The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease. Cell. Mol. Life Sci. 2009, 66, 565–583. [Google Scholar] [CrossRef]
- Zhou, H.; Blevins, M.A.; Hsu, J.Y.; Kong, D.; Galbraith, M.D.; Goodspeed, A.; Culp-Hill, R.; Oliphant, M.U.J.; Ramirez, D.; Zhang, L.; et al. Identification of a Small-Molecule Inhibitor That Disrupts the SIX1/EYA2 Complex, EMT, and Metastasis. Cancer Res. 2020, 80, 2689–2702. [Google Scholar] [CrossRef]
- Christensen, K.L.; Patrick, A.N.; McCoy, E.L.; Ford, H.L. The six family of homeobox genes in development and cancer. Adv. Cancer Res. 2008, 101, 93–126. [Google Scholar]
- Blevins, M.A.; Towers, C.G.; Patrick, A.N.; Zhao, R.; Ford, H.L. The SIX1-EYA transcriptional complex as a therapeutic target in cancer. Exp. Opin. Ther. Targets 2015, 19, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Rebay, I. Multiple Functions of the Eya Phosphotyrosine Phosphatase. Mol. Cell. Biol. 2016, 36, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Oghi, K.A.; Zhang, J.; Krones, A.; Bush, K.T.; Glass, C.K.; Nigam, S.K.; Aggarwal, A.K.; Maas, R.; Rose, D.W.; et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 2003, 426, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Rayapureddi, J.P.; Kattamuri, C.; Steinmetz, B.D.; Frankfort, B.J.; Ostrin, E.J.; Mardon, G.; Hegde, R.S. Eyes absent represents a class of protein tyrosine phosphatases. Nature 2003, 426, 295–298. [Google Scholar] [CrossRef]
- Tootle, T.L.; Silver, S.J.; Davies, E.L.; Newman, V.; Latek, R.R.; Mills, I.A.; Selengut, J.D.; Parlikar, B.E.; Rebay, I. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature 2003, 426, 299–302. [Google Scholar] [CrossRef]
- Pandey, R.N.; Rani, R.; Yeo, E.J.; Spencer, M.; Hu, S.; Lang, R.A.; Hegde, R.S. The Eyes Absent phosphatase-transactivator proteins promote proliferation, transformation, migration, and invasion of tumor cells. Oncogene 2010, 29, 3715–3722. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Jung, S.K.; Jeong, D.G.; Chung, S.J.; Kim, J.H.; Park, B.C.; Tonks, N.K.; Ryu, S.E.; Kim, S.J. Crystal structure of ED-Eya2: Insight into dual roles as a protein tyrosine phosphatase and a transcription factor. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 560–569. [Google Scholar] [CrossRef][Green Version]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef]
- Krishnan, N.; Jeong, D.G.; Jung, S.K.; Ryu, S.E.; Xiao, A.; Allis, C.D.; Kim, S.J.; Tonks, N.K. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J. Biol. Chem. 2009, 284, 16066–16070. [Google Scholar] [CrossRef]
- Liu, Y.; Long, Y.H.; Wang, S.Q.; Li, Y.F.; Zhang, J.H. Phosphorylation of H2A.X(T)(yr39) positively regulates DNA damage response and is linked to cancer progression. FEBS J. 2016, 283, 4462–4473. [Google Scholar] [CrossRef]
- Yuan, B.; Cheng, L.; Chiang, H.C.; Xu, X.; Han, Y.; Su, H.; Wang, L.; Zhang, B.; Lin, J.; Li, X.; et al. A phosphotyrosine switch determines the antitumor activity of ERbeta. J. Clin. Investig. 2014, 124, 3378–3390. [Google Scholar] [CrossRef] [PubMed]
- Mentel, M.; Ionescu, A.E.; Puscalau-Girtu, I.; Helm, M.S.; Badea, R.A.; Rizzoli, S.O.; Szedlacsek, S.E. WDR1 is a novel EYA3 substrate and its dephosphorylation induces modifications of the cellular actin cytoskeleton. Sci. Rep. 2018, 8, 2910. [Google Scholar] [CrossRef] [PubMed]
- Krueger, A.B.; Drasin, D.J.; Lea, W.A.; Patrick, A.N.; Patnaik, S.; Backos, D.S.; Matheson, C.J.; Hu, X.; Barnaeva, E.; Holliday, M.J.; et al. Allosteric inhibitors of the Eya2 phosphatase are selective and inhibit Eya2-mediated cell migration. J. Biol. Chem. 2014, 289, 16349–16361. [Google Scholar] [CrossRef] [PubMed]
- Krueger, A.B.; Dehdashti, S.J.; Southall, N.; Marugan, J.J.; Ferrer, M.; Li, X.; Ford, H.L.; Zheng, W.; Zhao, R. Identification of a selective small-molecule inhibitor series targeting the eyes absent 2 (Eya2) phosphatase activity. J. Biomol. Screen. 2013, 18, 85–96. [Google Scholar] [CrossRef]
- Rudmann, D.G. On-target and off-target-based toxicologic effects. Toxicol. Pathol. 2013, 41, 310–314. [Google Scholar] [CrossRef]
- Shraga, A.; Olshvang, E.; Davidzohn, N.; Khoshkenar, P.; Germain, N.; Shurrush, K.; Carvalho, S.; Avram, L.; Albeck, S.; Unger, T.; et al. Covalent Docking Identifies a Potent and Selective MKK7 Inhibitor. Cell Chem. Biol. 2019, 26, 98–108. [Google Scholar] [CrossRef]
- Chang, R.L.; Xie, L.; Xie, L.; Bourne, P.E.; Palsson, B.Ø. Drug Off-Target Effects Predicted Using Structural Analysis in the Context of a Metabolic Network Model. PLoS Comput. Biol. 2010, 6, e1000938. [Google Scholar] [CrossRef]
- Xie, L.; Xie, L.; Bourne, P.E. Structure-based systems biology for analyzing off-target binding. Curr. Opin. Struct. Biol. 2011, 21, 189–199. [Google Scholar] [CrossRef]
- Moumbock, A.F.A.; Li, J.; Mishra, P.; Gao, M.; Günther, S. Current computational methods for predicting protein interactions of natural products. Comput. Struct. Biotechnol. J. 2019, 17, 1367–1376. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Kang, C. Mechanisms of Action for Small Molecules Revealed by Structural Biology in Drug Discovery. Int. J. Mol. Sci. 2020, 21, 5262. https://doi.org/10.3390/ijms21155262
Li Q, Kang C. Mechanisms of Action for Small Molecules Revealed by Structural Biology in Drug Discovery. International Journal of Molecular Sciences. 2020; 21(15):5262. https://doi.org/10.3390/ijms21155262
Chicago/Turabian StyleLi, Qingxin, and CongBao Kang. 2020. "Mechanisms of Action for Small Molecules Revealed by Structural Biology in Drug Discovery" International Journal of Molecular Sciences 21, no. 15: 5262. https://doi.org/10.3390/ijms21155262
APA StyleLi, Q., & Kang, C. (2020). Mechanisms of Action for Small Molecules Revealed by Structural Biology in Drug Discovery. International Journal of Molecular Sciences, 21(15), 5262. https://doi.org/10.3390/ijms21155262