Tuning of Molecular Electrostatic Potential Enables Efficient Charge Transport in Crystalline Azaacenes: A Computational Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
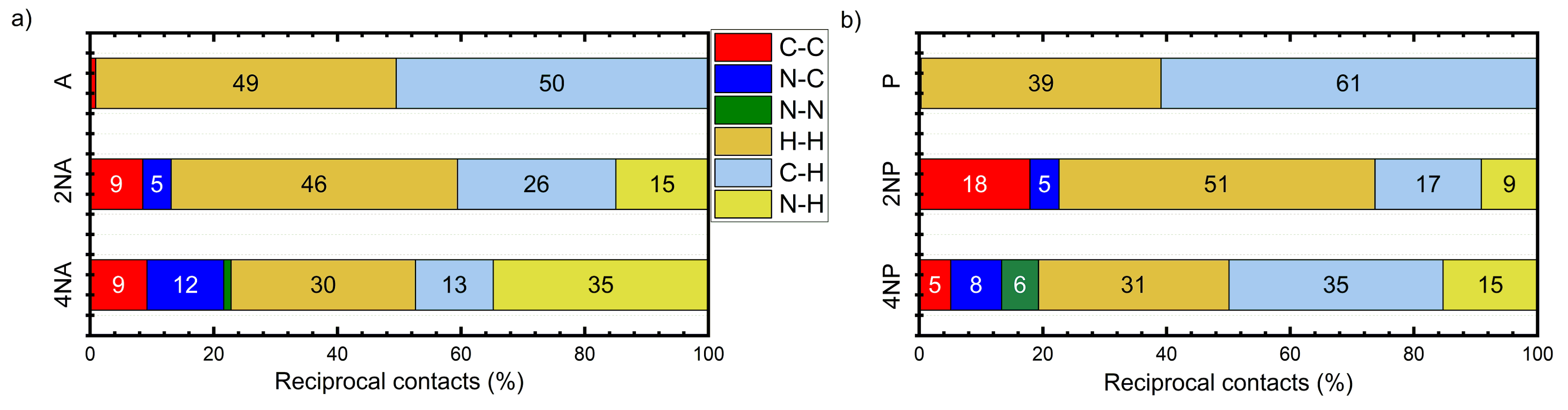
2. Results
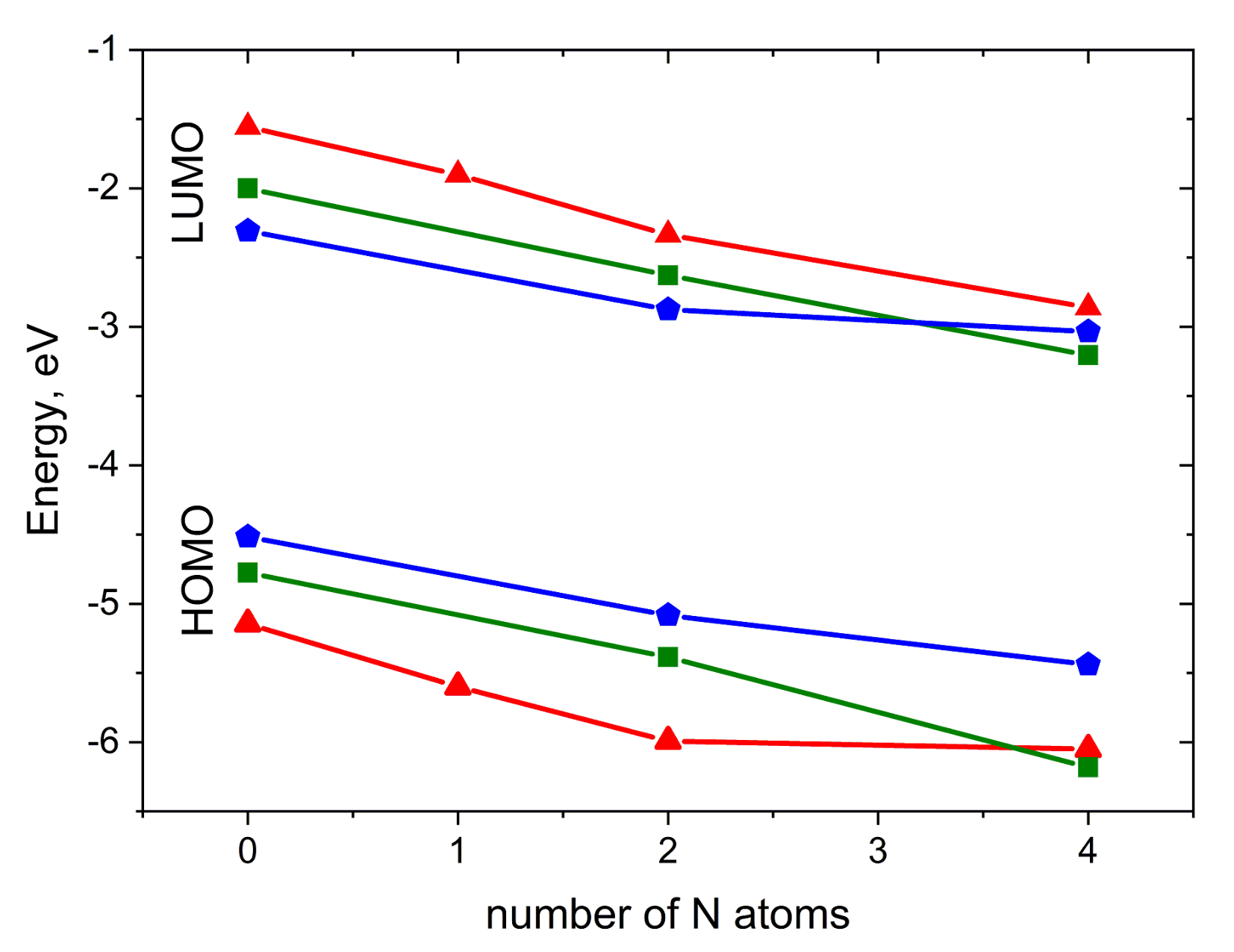
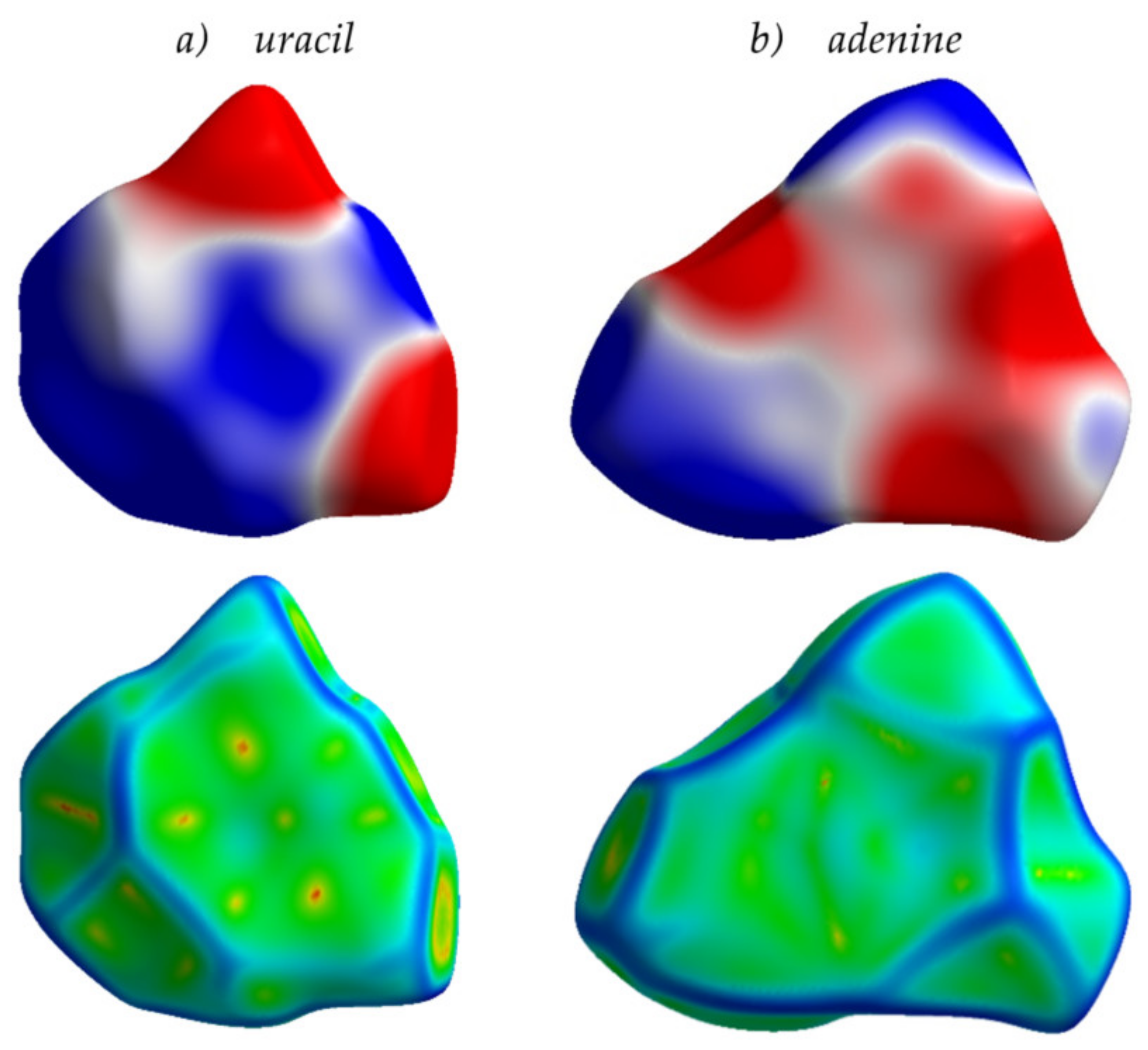
2.1. Molecules
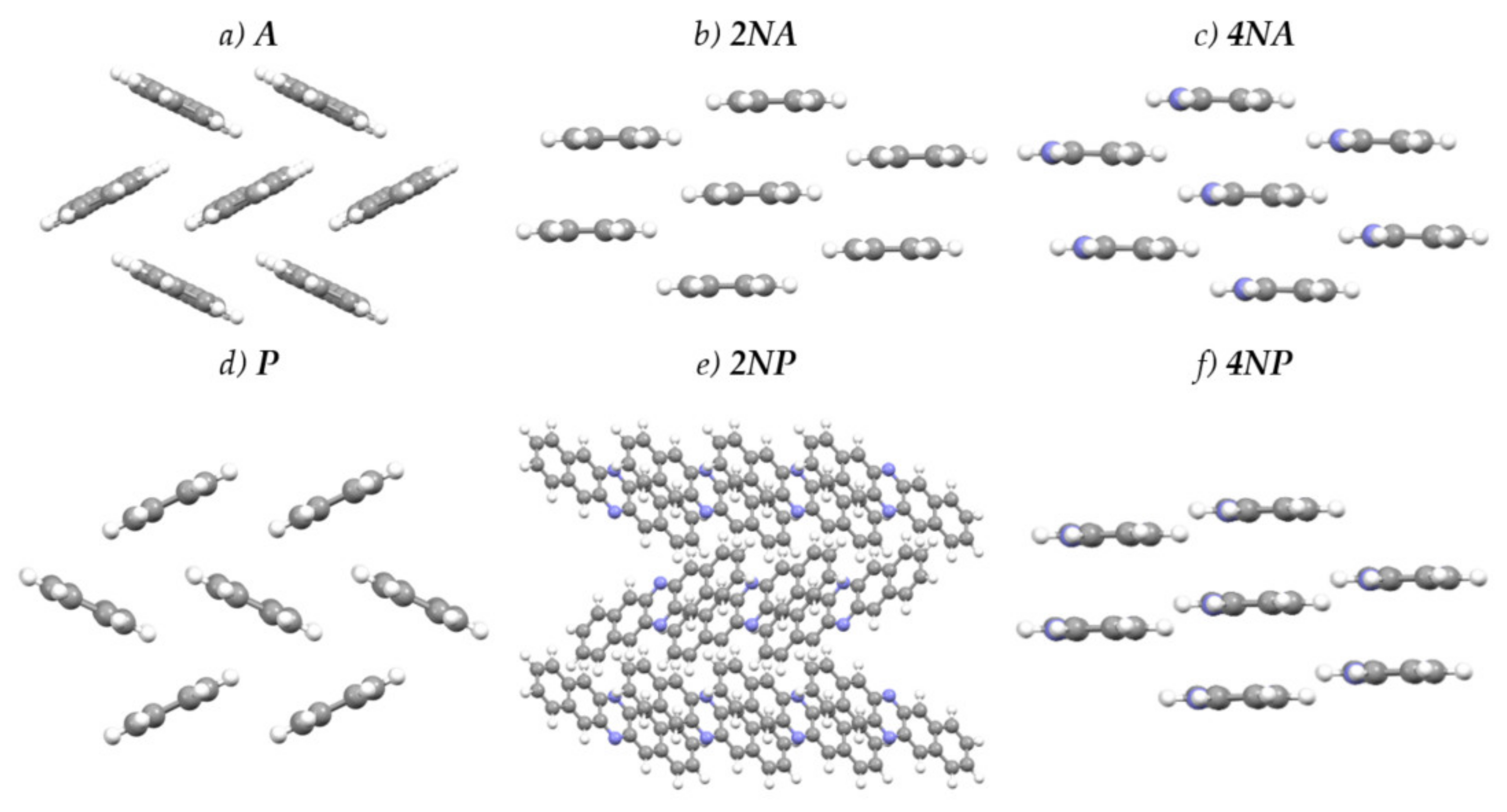
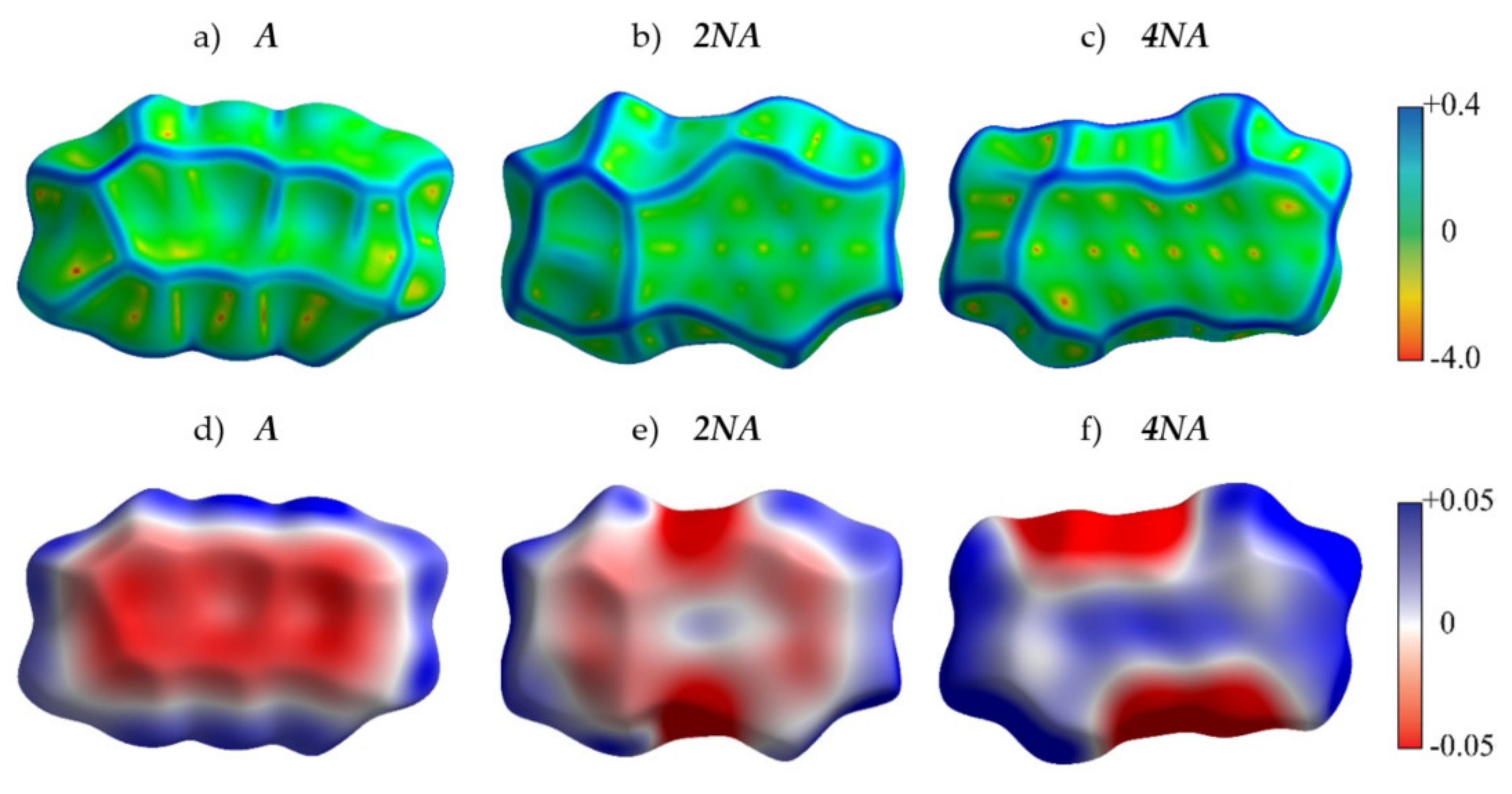
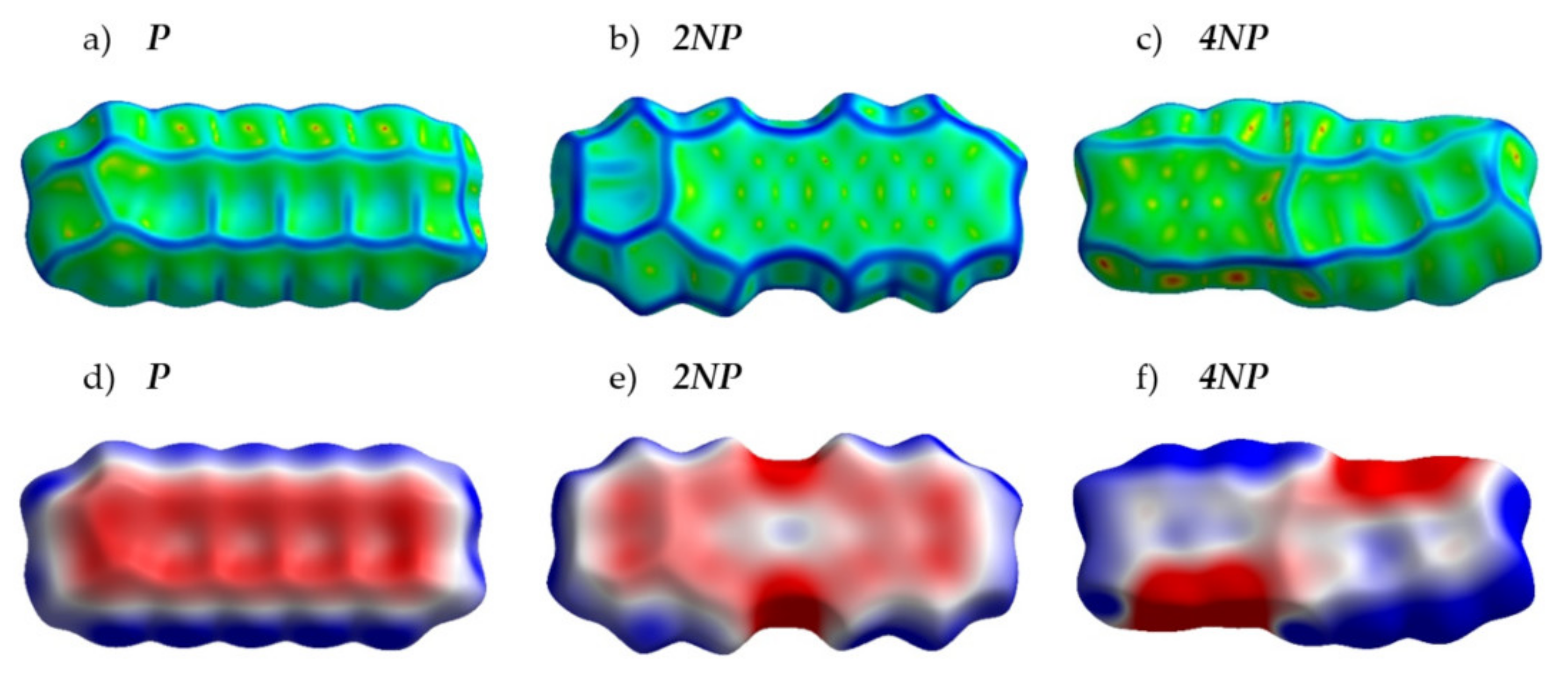
2.2. Crystals
2.3. Charge Transport
2.3.1. Reorganization Energies
2.3.2. Charge Transfer Integrals
2.3.3. Charge Mobilities
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schweicher, G.; Garbay, G.; Jouclas, R.; Vibert, F.; Devaux, F.; Geerts, Y.H. Molecular Semiconductors for Logic Operations: Dead-End or Bright Future? Adv. Mater. 2020, 32, 1905909. [Google Scholar] [CrossRef] [PubMed]
- Ostroverkhova, O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Osuka, A.; Fukuzumi, S.; Kandori, H.; Aso, Y. Chemical Science of π-Electron Systems; Springer: Tokyo, Japan, 2015. [Google Scholar]
- Schweicher, G.; Olivier, Y.; Lemaur, V.; Geerts, Y.H. What Currently Limits Charge Carrier Mobility in Crystals of Molecular Semiconductors? Isr. J. Chem. 2014, 54, 595–620. [Google Scholar] [CrossRef]
- Troisi, A. Charge transport in high mobility molecular semiconductors: Classical models and new theories. Chem. Soc. Rev. 2011, 40, 2347–2358. [Google Scholar] [CrossRef]
- Köhler, A.; Bässler, H. Electronic Processes in Organic Semiconductors: An Introduction; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Li, Y.; Coropceanu, V.; Brédas, J.-L. Electronics: Organic Semiconductors. In The WSPC Reference on Organic; Brédas, J.-L., Marder, S.R., Eds.; World Scientific: Singapore, 2016; Chapter 7; p. 193. [Google Scholar]
- Sosorev, A.Y. Simple charge transport model for efficient search of high-mobility organic semiconductor crystals. Mater. Des. 2020, 192, 108730. [Google Scholar] [CrossRef]
- Sosorev, A.Y. Role of intermolecular charge delocalization and its dimensionality in efficient band-like electron transport in crystalline 2,5-difluoro-7,7,8,8-tetracyanoquinodimethane (F2-TCNQ). Phys. Chem. Chem. Phys. 2017, 19, 25478–25486. [Google Scholar] [CrossRef] [Green Version]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Brédas, J.L. Charge Transport in Organic Semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef]
- Corpinot, M.K.; Bučar, D.-K. A Practical Guide to the Design of Molecular Crystals. Cryst. Growth Des. 2019, 19, 1426–1453. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y.; Chao, I. Toward the Rational Design of Functionalized Pentacenes: Reduction of the Impact of Functionalization on the Reorganization Energy. Chem. Phys. Chem. 2006, 7, 2003–2007. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.E.; Yang, J.; Day, G.M. Predicted energy–structure–function maps for the evaluation of small molecule organic semiconductors. J. Mater. Chem. C 2017, 5, 7574. [Google Scholar] [CrossRef] [Green Version]
- Klues, M.; Witte, G. Crystalline packing in pentacene-like organic semiconductors. CrystEngComm 2018, 20, 63. [Google Scholar] [CrossRef]
- Chernyshov, I.Y.; Vener, M.V.; Feldman, E.V.; Paraschuk, D.Y.; Sosorev, A.Y. Inhibiting low-frequency vibrations explains exceptionally high electron mobility in 2,5-difluoro-7,7,8,8-tetracyanoquinodimethane (F2-TCNQ) single crystals. J. Phys. Chem. Lett. 2017, 8, 2875–2880. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P.; Tong, Q.; Untenecker, H. Crystal design using multipolar electrostatic interactions: A concept study for organic electronics. Beilstein J. Org. Chem. 2013, 9, 2367–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Dong, H.; Hu, W.; Liu, Y.; Zhu, D. Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics. Chem. Rev. 2012, 112, 2208–2267. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Chen, Y.-D.; Chen, C.-H.; Wen, Y.-S.; Lin, J.T.; Chen, H.-Y.; Kuo, M.-Y.; Chao, I. Crystal Engineering for π-π Stacking via Interaction between Electron-Rich and Electron-Deficient Heteroaromatics. J. Org. Chem. 2008, 73, 4608–4614. [Google Scholar] [CrossRef]
- Dou, J.-H.; Zheng, Y.-Q.; Yao, Z.-F.; Yu, Z.-A.; Lei, T.; Shen, X.; Luo, X.-Y.; Sun, J.; Zhang, S.-D.; Ding, Y.-F.; et al. Fine-Tuning of Crystal Packing and Charge Transport Properties of BDOPV Derivatives through Fluorine Substitution. J. Am. Chem. Soc. 2015, 137, 15947–15956. [Google Scholar] [CrossRef]
- Menard, E.; Podzorov, V.; Hur, S.H.; Gaur, A.; Gershenson, M.E.; Rogers, J.A. High-Performance n- and p-Type Single-Crystal Organic Transistors with Free-Space Gate Dielectrics. Adv. Mater. 2004, 16, 2097–2101. [Google Scholar] [CrossRef]
- Krupskaya, Y.; Gibertini, M.; Marzari, N.; Morpurgo, A.F. Band-like electron transport with record-high mobility in the TCNQ family. Adv. Mater. 2015, 27, 2453–2458. [Google Scholar] [CrossRef] [Green Version]
- Komissarova, E.A.; Dominsky, D.I.; Zhulanov, V.E.; Abashev, G.G.; Siddiqui, A.S.; Singh, S.P.; Sosorev, A.Y.; Paraschuk, D.Y. Unraveling the unusual effect of fluorination on crystal packing in organic semiconductor. Phys. Chem. Chem. Phys. 2020, 22, 1665–1673. [Google Scholar] [CrossRef]
- Lin, P.-P.; Qin, G.-Y.; Zhang, N.-X.; Fan, J.-X.; Hao, X.-L.; Zou, L.-Y.; Ren, A.-M. The roles of heteroatoms and substituents on the molecular packing motif from herringbone to π-stacking: A theoretical study on electronic structures and intermolecular interaction of pentacene derivatives. Org. Electron. 2020, 78, 105606. [Google Scholar] [CrossRef]
- Maly, K.E. Acenes vs N-Heteroacenes: The Effect of N-Substitution on the Structural Features of Crystals of Polycyclic Aromatic Hydrocarbons. Cryst. Growth Des. 2011, 11, 5628–5633. [Google Scholar] [CrossRef]
- Chen, X.-K.; Guo, J.-F.; Zou, L.-Y.; Ren, A.-M.; Fan, J.-X. A Promising Approach to Obtain Excellent n-Type OrganicField-Effect Transistors—Introducing Pyrazine Ring. J. Phys. Chem. C 2011, 115, 21416–21428. [Google Scholar] [CrossRef]
- Valeev, E.F.; Coropceanu, V.; da Silva Filho, D.A.; Salman, S.; Bredas, J.-L. Effect of Electronic Polarization on Charge-Transport Parameters in Molecular Organic Semiconductors. J. Am. Chem. Soc. 2006, 128, 9882. [Google Scholar] [CrossRef]
- Miao, Q. Ten Years of N-Heteropentacenes as Semiconductors for Organic Thin-Film Transistors. Adv. Mater. 2014, 26, 5541–5549. [Google Scholar] [CrossRef] [PubMed]
- Reese, C.; Chung, W.-J.; Ling, M.-M.; Roberts, M.; Bao, Z. High-performance microscale single-crystal transistors by lithography on an elastomer dielectric Appl. Phys. Lett. 2006, 89, 202108. [Google Scholar]
- Brédas, J.-L.; Marder, S.R.; André, J.-M. Electronics: Organic Semiconductors. In The WSPC Reference on Organic; Brédas, J.-L., Marder, S.R., Eds.; World Scientific: Singapore, 2016; Chapter 1; p. 1. [Google Scholar]
- Winkler, M.; Houk, K. Nitrogen-Rich Oligoacenes: Candidates for n-Channel Organic Semiconductors. J. Am. Chem. Soc. 2007, 129, 1805. [Google Scholar] [CrossRef]
- Appleton, A.L.; Brombosz, S.M.; Barlow, S.; Sears, J.S.; Brédas, J.-L.; Marder, S.R.; Bunz, U.H.F. Effects of electronegative substitution on the optical and electronic properties of acenes and diazaacenes. Nat. Commun. 2010, 1, 91. [Google Scholar] [CrossRef]
- Dhar, J.; Salzner, U.; Patil, S. Trends in molecular design strategies for ambient stable n-channel organic field effect transistors. J. Mater. Chem. C 2017, 5, 7404. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel Tools for Visualizing and Exploring Intermolecular Interactions in Molecular Crystals. Acta Crystallogr. Sect. B 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215. [Google Scholar] [CrossRef]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Kato, T.; Yamabe, T. Electron−Phonon Interactions in the Monoanions of Polycyanodienes. J. Phys. Chem. A 2004, 108, 11223–11233. [Google Scholar] [CrossRef]
- Gümüş, S.; Akbay, M. A Computational Study on Substituted and Unsubstituted Mono and Diazaanthracenes. Polycycl. Aromat. Compd. 2013, 33, 519–532. [Google Scholar] [CrossRef]
- Chen, W.-C.; Chao, I. Molecular Orbital-Based Design of π-Conjugated Organic Materials with Small Internal Reorganization Energy: Generation of Nonbonding Character in Frontier Orbitals. J. Phys. Chem. C 2014, 118, 20176–20183. [Google Scholar] [CrossRef]
- Myers, A.B. Resonance Raman Intensities and Charge-Transfer Reorganization Energies. Chem. Rev. 1996, 96, 911–926. [Google Scholar] [CrossRef]
- Sosorev, A.Y. The Electron-Vibrational Interaction in a Thiophene—Phenylene Co-oligomer and Its Relationship to the Raman Spectrum. Mosc. Univ. Phys. Bull. 2019, 74, 639–645. [Google Scholar] [CrossRef]
- Scholz, R.; Gisslén, L.; Himcinschi, C.; Calzado, E.M.; Louis, E.; San Fabián Maroto, E.; Díaz-García, M.A. Asymmetry between Absorption and Photoluminescence Line Shapes of TPD: Spectroscopic Fingerprint of the Twisted Biphenyl Core. J. Phys. Chem. A 2009, 113, 315. [Google Scholar] [CrossRef]
- Roncali, J.; Leriche, P.; Cravino, A. From One- to Three-Dimensional Organic Semiconductors: In Search of the Organic Silicon. Adv. Mater. 2007, 19, 2045–2060. [Google Scholar] [CrossRef] [Green Version]
- Sosorev, A.Y.; Trukhanov, V.A.; Maslennikov, D.R.; Borshchev, O.V.; Polyakov, R.A.; Skorotetcky, M.S.; Surin, N.M.; Kazantsev, M.S.; Dominskiy, D.I.; Tafeenko, V.A.; et al. Paraschuk, Fluorinated Thiophene-Phenylene Co-Oligomers for Optoelectronic Devices. ACS Appl. Mater. Interfaces 2020, 12, 9507–9519. [Google Scholar] [CrossRef]
- Kotadiya, N.B.; Mondal, A.; Blom, P.W.M.; Andrienko, D.; Wetzelaer, G.-J.A.H. A window to trap-free charge transport in organic semiconducting thin films. Nat. Mater. 2019, 18, 1182–1186. [Google Scholar] [CrossRef]
- Ukwitegetse, N.; Saris, P.J.G.; Sommer, J.R.; Haiges, R.M.; Djurovich, P.I.; Thompson, M.E. Tetra-Aza-Pentacenes by means of a One-Pot Friedländer Synthesis Chem. Eur. J. 2019, 25, 1472. [Google Scholar] [CrossRef] [PubMed]
- Trukhanov, V.A.; Dominskiy, D.I.; Parashchuk, O.D.; Feldman, E.V.; Surin, N.M.; Svidchenko, E.A.; Skorotetcky, M.S.; Borshchev, O.V.; Paraschuk, D.Y.; Sosorev, A.Y. Exploring the impact of N-substitution on structural, electronic, optical and vibrational properties of thiophene-phenylene co-oligomer. RSC Adv. 2020, 10, 28128–28138. [Google Scholar] [CrossRef]
- Maia, F.F., Jr.; Freire, V.N.; Caetano, E.W.S.; Azevedo, D.L.; Sales, F.A.M.; Albuquerque, E.L. Anhydrous crystals of DNA bases are wide gap semiconductors. J. Chem. Phys. 2011, 134, 175101. [Google Scholar] [CrossRef]
- O’Brien, E.; Holt, M.E.; Thompson, M.K.; Salay, L.E.; Ehlinger, A.C.; Chazin, W.J.; Barton, J.K. The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport. Science 2017, 355, eaag1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, K.J.; Chemler, J.A.; Bartels, P.L.; O’Brien, E.; Marvin, M.L.; Ortega, J.; Stern, R.H.; Raskin, L.; Li, G.-M.; Sherman, D.H.; et al. A human MUTYH variant linking colonic polyposis to redox degradation of the [4Fe4S]2+ cluster. Nat. Chem. 2018, 10, 873–880. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Gordon, M.S.; Schmidt, M.W. Advances in Electronic Structure Theory: GAMESS a Decade Later. In Theory and Applications of Computational Chemistry: The First Forty Years; Dykstra, C.E., Frenking, G., Kim, K.S., Scuseria, G.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 1167–1189. [Google Scholar]
- Sosorev, A.Y.; Chernyshov, I.Y.; Paraschuk, D.Y.; Vener, M.V. Intra- and Intermolecular Vibrations of Organic Semiconductors and Their Role in Charge Transport. In Molecular Spectroscopy; Ozaki, Y., Wójcik, M.J., Popp, J., Eds.; Wiley: Hoboken, NJ, USA, 2019; Chapter 15. [Google Scholar]
- Baumeier, B.; Kirkpatrick, J.; Andrienko, D. Density-functional based determination of intermolecular charge transfer properties for large-scale morphologies. Phys. Chem. Chem. Phys. 2010, 12, 11103. [Google Scholar] [CrossRef]
- Kirkpatrick, J. An Approximate Method for Calculating Transfer Integrals Based on the ZINDO Hamiltonian. Int. J. Quantum Chem. 2008, 108, 51. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Kobayashi, N.; Hosoi, S.; Koshitani, N.; Murakami, D.; Shirasawa, R.; Kudo, Y.; Hobara, D.; Tokita, Y.; Itabashi, M. Hopping and band mobilities of pentacene, rubrene, and 2,7-dioctyl[1]benzothieno[3,2-b][1]benzothiophene (C8-BTBT) from first principle calculations. J. Chem. Phys. 2013, 139, 8. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17, University of Western Australia. 2017. Available online: http://hirshfeldsurface.net (accessed on 6 August 2020).
- Turner, M.J.; Grabowsky, S.; Jayatilaka, D.; Spackman, M.A. Accurate and Efficient Model Energies for Exploring Intermolecular Interactions in Molecular Crystals. J. Phys. Chem. Lett. 2014, 5, 4249. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer model energies and energy frameworks: Extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sosorev, A.; Dominskiy, D.; Chernyshov, I.; Efremov, R. Tuning of Molecular Electrostatic Potential Enables Efficient Charge Transport in Crystalline Azaacenes: A Computational Study. Int. J. Mol. Sci. 2020, 21, 5654. https://doi.org/10.3390/ijms21165654
Sosorev A, Dominskiy D, Chernyshov I, Efremov R. Tuning of Molecular Electrostatic Potential Enables Efficient Charge Transport in Crystalline Azaacenes: A Computational Study. International Journal of Molecular Sciences. 2020; 21(16):5654. https://doi.org/10.3390/ijms21165654
Chicago/Turabian StyleSosorev, Andrey, Dmitry Dominskiy, Ivan Chernyshov, and Roman Efremov. 2020. "Tuning of Molecular Electrostatic Potential Enables Efficient Charge Transport in Crystalline Azaacenes: A Computational Study" International Journal of Molecular Sciences 21, no. 16: 5654. https://doi.org/10.3390/ijms21165654