Enforced C-Src Activation Causes Compartmental Dysregulation of PI3K and PTEN Molecules in Lipid Rafts of Tongue Squamous Carcinoma Cells by Attenuating Rac1-Akt-GLUT-1-Mediated Sphingolipid Synthesis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. GA Inhibits TSC Cell Invasion by Downregulating MMP-2 and -9 Expression
2.2. GA Decreased Lipid Raft Membrane Targeting of PI3K, Rac1, Akt, and GLUT-1 Molecules
2.3. Perturbation of the Lipid Raft Membrane Targeting of PI3k-Rac1-Akt-Glut-1 Signaling Molecules and the Formation of P85α-Unphosphorylated PTEN Complexes Via the Inductive Effect of GA on the Formation of Inactive Rac1-P-Ck2α (Tyr 255) Complexes
2.4. Formation of Inactive Rac1-p-CK2α (Tyr 255) Complexes by GA-Induced C-Src Activation Attenuates Akt-Mediated Glut-1 Membrane Localization, and Suppresses MMP-2/-9-Mediated Cell Invasion
2.5. Impairment of Rac1 Activation by GA-Induced c-Src Activity Attenuates Akt-Regulated GLUT-1-Mediated Membrane Lipid Homeostasis, Leading to Inhibition of Cell Invasion
2.6. GA-Induced Rac1 Inactivation Was Essential in Suppressing NF-κB-Mediated MMP-2/-9 Expression
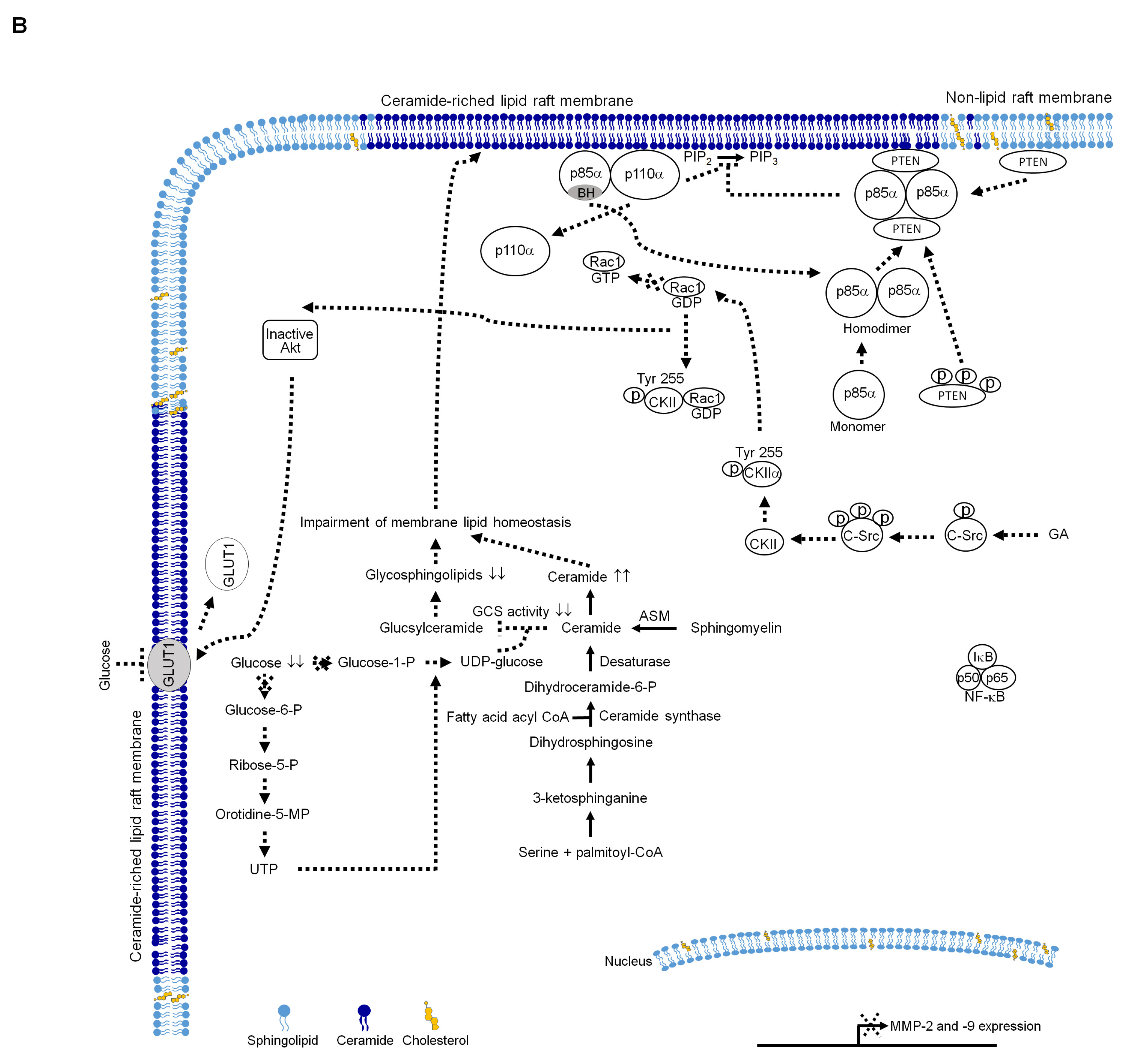
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals, Reagents, and Plasmids
4.3. Antibodies
4.4. Cell Viability Assay
4.5. Determination of Caspase-3 Activity
4.6. RT-PCR
4.7. Density-Based Membrane Flotation Technique
4.8. Western Blot and Co-Immunoprecipitation
4.9. Cell Surface Biotinylation
4.10. Rac1 Activation Assay
4.11. Measurement of Intracellular Glucose
4.12. Determination of GCS Activity
4.13. Determination of Cholesterol, Sphingolipid, and Ceramide
4.14. Measurements of MMP-2 and -9 Promoter Activities
4.15. In Vitro Invasion Assay
4.16. Gelatin Zymography
4.17. Plasmid, siRNA, and shRNA Transfection
4.18. NF-κB Promoter Activity
4.19. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASM | Acid sphingomyelinase |
| CK2 | Casein kinase 2 |
| GLUT-1 | Glucose transporter-1 |
| MMP-2 | Matrix metalloproteinase-2 |
| MMP-2 | Matrix metalloproteinase-9 |
| NF-κB | Nuclear factor-kappa B |
| PTEN | Phosphatase and tensin homolog deleted from chromosome 10 |
| PI3K | Phosphatidylinositol 3-kinase |
| PIP2 | Phosphatidylinositol-4,5-bisphosphate |
| PIP3 | Phosphatidylinositol-3,4,5-trisphosphate |
| Akt | Protein kinase B |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| shRNA | Short hairpin RNA |
| SP-1 | Stimulatory protein-1 |
| TSC | Tongue squamous carcinoma |
References
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K.; Weissman, J.S. Membranes in Balance: Mechanisms of Sphingolipid Homeostasis. Mol. Cell 2010, 40, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.L.; Wang, S.G.; Chan, W.L.; Lee, C.H.; Wu, T.S.; Lin, M.L.; Chen, S.S. Impairment of Membrane Lipid Homeostasis by Bichalcone Analog TSWU-BR4 Attenuates Function of GRP78 in Regulation of the Oxidative Balance and Invasion of Cancer Cells. Cells 2020, 9, 371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payapilly, A.; Malliri, A. Compartmentalisation of RAC1 signalling. Curr. Opin. Cell Biol. 2018, 54, 50–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcaro, A.; Aubert, M.; Espinosa del Hierro, M.E.; Khanzada, U.K.; Angelidou, S.; Tetley, T.D.; Bittermann, A.G.; Frame, M.C.; Seckl, M.J. Critical role for lipid raft-associated Src kinases in activation of PI3K-Akt signalling. Cell. Signal. 2007, 19, 1081–1092. [Google Scholar] [CrossRef]
- Stambolic, V.; Suzuki, A.; De la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Cheung, L.W.; Walkiewicz, K.W.; Besong, T.M.; Guo, H.; Hawke, D.H.; Arold, S.T.; Mills, G.B. Regulation of the PI3K pathway through a p85alpha monomer-homodimer equilibrium. Elife 2015, 4, e06866. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.S.; Jung, J.E.; Niizuma, K.; Chan, P.H. CK2 is a novel negative regulator of NADPH oxidase and a neuroprotectant in mice after cerebral ischemia. J. Neurosci. 2009, 29, 14779–14789. [Google Scholar] [CrossRef]
- Donella-Deana, A.; Cesaro, L.; Sarno, S.; Ruzzene, M.; Brunati, A.M.; Marin, O.; Vilk, G.; Doherty-Kirby, A.; Lajoie, G.; Litchfield, D.W.; et al. Tyrosine phosphorylation of protein kinase CK2 by Src-related tyrosine kinases correlates with increased catalytic activity. Biochem. J. 2003, 372, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Trembley, J.H.; Wang, G.; Unger, G.; Slaton, J.; Ahmed, K. Protein kinase CK2 in health and disease: CK2: A key player in cancer biology. Cell. Mol. Life Sci. 2009, 66, 1858–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.A.; Barry, D.A.; Leggett, R.W.; Mueller, C.R. Casein kinase II-mediated phosphorylation of the C terminus of Sp1 decreases its DNA binding activity. J. Biol. Chem. 1997, 272, 13489–13495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipiak, K.; Kubinski, K.; Hellman, U.; Ramos, A.; De Pascual-Teresa, B. Human protein kinase CK2 phosphorylates matrix metalloproteinase 2 and inhibits its activity. Chembiochem 2014, 15, 1873–1876. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Seiki, M. Regulatory mechanism of 92 kDa type IV collagenase gene expression which is associated with invasiveness of tumor cells. Oncogene 1993, 8, 395–405. [Google Scholar] [PubMed]
- Li, Z.; Guo, Y.; Jiang, H.; Zhang, T.; Jin, C.; Young, C.Y.; Yuan, H. Differential regulation of MMPs by E2F1, Sp1 and NF-kappa B controls the small cell lung cancer invasive phenotype. BMC Cancer 2014, 14, 276. [Google Scholar] [CrossRef] [Green Version]
- Philip, S.; Bulbule, A.; Kundu, G.C. Matrix metalloproteinase-2: Mechanism and regulation of NF-kappaB-mediated activation and its role in cell motility and ECM-invasion. Glycoconj. J. 2004, 21, 429–941. [Google Scholar] [CrossRef]
- Bond, M.; Fabunmi, R.P.; Baker, A.H.; Newby, A.C. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: An absolute requirement for transcription factor NF-kappa B. FEBS Lett. 1998, 435, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, B.B. Nuclear factor-kappaB: The enemy within. Cancer Cell 2004, 6, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Orlowski, R.Z.; Baldwin, A.S., Jr. NF-kappaB as a therapeutic target in cancer. Trends Mol. Med. 2002, 8, 385–389. [Google Scholar] [CrossRef] [Green Version]
- Ikebe, T.; Shinohara, M.; Takeuchi, H.; Beppu, M.; Kurahara, S.; Nakamura, S.; Shirasuna, K. Gelatinolytic activity of matrix metalloproteinase in tumor tissues correlates with the invasiveness of oral cancer. Clin. Exp. Metastasis 1999, 17, 315–323. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.Y.; Yu, F.X.; Luo, Y.; Hagen, T. Oncogenic activation of the PI3K/Akt pathway promotes cellular glucose uptake by downregulating the expression of thioredoxin-interacting protein. Cell. Signal. 2016, 28, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Ishii, G.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avanzato, D.; Pupo, E.; Ducano, N.; Isella, C.; Bertalot, G.; Luise, C.; Pece, S.; Bruna, A.; Rueda, O.M.; Caldas, C.; et al. High USP6NL Levels in Breast Cancer Sustain Chronic AKT Phosphorylation and GLUT1 Stability Fueling Aerobic Glycolysis. Cancer Res. 2018, 78, 3432–3444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, L.; Ito, M.; Zhang, G.; Hirabayashi, Y.; Inokuchi, J.; Yamagata, T. Effects of endoglycoceramidase or D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol on glucose uptake, glycolysis, and mitochondrial respiration in HL60 cells. Arch. Biochem. Biophys. 1998, 359, 107–114. [Google Scholar] [CrossRef]
- Stathem, M.; Marimuthu, S.; O’Neal, J.; Rathmell, J.C.; Chesney, J.A.; Beverly, L.J.; Siskind, L.J. Glucose availability and glycolytic metabolism dictate glycosphingolipid levels. J. Cell. Biochem. 2015, 116, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Yu, J.; Pohorly, J.E.; Kakuda, Y. Polyphenolics in grape seeds-biochemistry and functionality. J. Med. Food 2003, 6, 291–299. [Google Scholar] [CrossRef]
- Zuo, Y.; Chen, H.; Deng, Y. Simultaneous determination of catechins, caffeine and gallic acids in green, Oolong, black and pu-erh teas using HPLC with a photodiode array detector. Talanta 2002, 57, 307–316. [Google Scholar] [CrossRef]
- Liao, C.C.; Chen, S.C.; Huang, H.P.; Wang, C.J. Gallic acid inhibits bladder cancer cell proliferation and migration via regulating fatty acid synthase (FAS). J. Food Drug Anal. 2018, 26, 620–627. [Google Scholar] [CrossRef]
- Jafari, N.; Drury, J.; Morris, A.J.; Onono, F.O.; Stevens, P.D.; Gao, T.; Liu, J.; Wang, C.; Lee, E.Y.; Weiss, H.L.; et al. De Novo Fatty Acid Synthesis-Driven Sphingolipid Metabolism Promotes Metastatic Potential of Colorectal Cancer. Mol. Cancer Res. 2019, 17, 140–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.L.; Chen, S.S. Activation of Casein Kinase II by Gallic Acid Induces BIK-BAX/BAK-Mediated ER Ca(++)-ROS-Dependent Apoptosis of Human Oral Cancer Cells. Front. Physiol. 2017, 8, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Johnson, K.R. Cadherins as modulators of cellular phenotype. Annu. Rev. Cell Dev. Biol. 2003, 19, 207–235. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lowry, P.R.; Zhou, X.; Depry, C.; Wei, Z.; Wong, G.W.; Zhang, J. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc. Natl. Acad. Sci. USA 2011, 108, 14509–14514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef] [Green Version]
- Eich, C.; Manzo, C.; De Keijzer, S.; Bakker, G.J.; Reinieren-Beeren, I.; Garcia-Parajo, M.F.; Cambi, A. Changes in membrane sphingolipid composition modulate dynamics and adhesion of integrin nanoclusters. Sci. Rep. 2016, 6, 20693. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Wada, R.; Sasaki, T.; Deng, C.; Bierfreund, U.; Sandhoff, K.; Proia, R.L. A vital role for glycosphingolipid synthesis during development and differentiation. Proc. Natl. Acad. Sci. USA 1999, 96, 9142–9147. [Google Scholar] [CrossRef] [Green Version]
- Perona, R.; Montaner, S.; Saniger, L.; Sanchez-Perez, I.; Bravo, R.; Lacal, J.C. Activation of the nuclear factor-kappaB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997, 11, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Madrid, L.V.; Wang, C.Y.; Guttridge, D.C.; Schottelius, A.J.; Baldwin, A.S., Jr.; Mayo, M.W. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol. Cell. Biol. 2000, 20, 1626–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, H.; Chiba, H.; Miyoshi, H.; Sugita, T.; Toriumi, W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 1999, 274, 30353–30356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, C.Y.; Barberi, T.J.; Ghosh, P.; Longo, D.L. Phosphorylation of RelA/p65 on serine 536 defines an I{kappa}B{alpha}-independent NF-{kappa}B pathway. J. Biol. Chem. 2005, 280, 34538–34547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adli, M.; Baldwin, A.S. IKK-i/IKKepsilon controls constitutive, cancer cell-associated NF-kappaB activity via regulation of Ser-536 p65/RelA phosphorylation. J. Biol. Chem. 2006, 281, 26976–26984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattioli, I.; Sebald, A.; Bucher, C.; Charles, R.P.; Nakano, H.; Doi, T.; Kracht, M.; Schmitz, M.L. Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J. Immunol. 2004, 172, 6336–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardes, N.; Fialho, A.M. Perturbing the Dynamics and Organization of Cell Membrane Components: A New Paradigm for Cancer-Targeted Therapies. Int. J. Mol. Sci. 2018, 19, 3871. [Google Scholar] [CrossRef] [Green Version]
- Giussani, P.; Brioschi, L.; Bassi, R.; Riboni, L.; Viani, P. Phosphatidylinositol 3-kinase/AKT pathway regulates the endoplasmic reticulum to golgi traffic of ceramide in glioma cells: A link between lipid signaling pathways involved in the control of cell survival. J. Biol. Chem. 2009, 284, 5088–5096. [Google Scholar] [CrossRef] [Green Version]
- Schubert, K.M.; Scheid, M.P.; Duronio, V. Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J. Biol.Chem. 2000, 275, 13330–13335. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.M.; Feng, J.; Hemmings, B.A. Identification of a plasma membrane Raft-associated PKB Ser473 kinase activity that is distinct from ILK and PDK1. Curr. Biol. 2002, 12, 1251–1255. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Luo, Q.; Halim, A.; Song, G. Targeting lipid metabolism of cancer cells: A promising therapeutic strategy for cancer. Cancer Lett. 2017, 401, 39–45. [Google Scholar] [CrossRef]
- Zien, P.; Duncan, J.S.; Skierski, J.; Bretner, M.; Litchfield, D.W.; Shugar, D. Tetrabromobenzotriazole (TBBt) and tetrabromobenzimidazole (TBBz) as selective inhibitors of protein kinase CK2: Evaluation of their effects on cells and different molecular forms of human CK2. Biochim. Biophys. Acta 2005, 1754, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Ponce, D.P.; Maturana, J.L.; Cabello, P.; Yefi, R.; Niechi, I.; Silva, E.; Armisen, R.; Galindo, M.; Antonelli, M.; Tapia, J.C. Phosphorylation of AKT/PKB by CK2 is necessary for the AKT-dependent up-regulation of beta-catenin transcriptional activity. J. Cell. Physiol. 2011, 226, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Ponce, D.P.; Yefi, R.; Cabello, P.; Maturana, J.L.; Niechi, I.; Silva, E.; Galindo, M.; Antonelli, M.; Marcelain, K.; Armisen, R.; et al. CK2 functionally interacts with AKT/PKB to promote the beta-catenin-dependent expression of survivin and enhance cell survival. Mol. Cell. Biochem. 2011, 356, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Ishizawar, R.; Parsons, S.J. c-Src and cooperating partners in human cancer. Cancer Cell 2004, 6, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, C.A.; Kamps, M.P.; Meisler, A.I.; Pipas, J.M.; Eckhart, W. pp60c-src activation in human colon carcinoma. J. Clin. Investig. 1989, 83, 2025–2033. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, S.; Jiang, B.; Huang, L.; Ji, Z.; Li, X.; Zhou, H.; Han, A.; Chen, A.; Wu, Y.; et al. c-Src phosphorylation and activation of hexokinase promotes tumorigenesis and metastasis. Nat. Commun. 2017, 8, 13732. [Google Scholar] [CrossRef] [PubMed]
- Megha; London, E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): Implications for lipid raft structure and function. J. Biol. Chem. 2004, 279, 9997–10004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.L.; Lu, Y.C.; Chen, H.Y.; Lee, C.C.; Chung, J.G.; Chen, S.S. Suppressing the formation of lipid raft-associated Rac1/PI3K/Akt signaling complexes by curcumin inhibits SDF-1alpha-induced invasion of human esophageal carcinoma cells. Mol. Carcinog. 2014, 53, 360–379. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.L.; Chen, S.S.; Hsu, W.T.; Lu, Y.C.; Lee, C.C.; Wu, T.S.; Lin, M.L. Suppression of phospho-p85alpha-GTP-Rac1 lipid raft interaction by bichalcone analog attenuates cancer cell invasion. Mol. Carcinog. 2016, 55, 2106–2120. [Google Scholar] [CrossRef]
- Dobrowsky, R.T.; Kolesnick, R.N. Analysis of sphingomyelin and ceramide levels and the enzymes regulating their metabolism in response to cell stress. Methods Cell Biol. 2001, 66, 135–165. [Google Scholar] [PubMed]

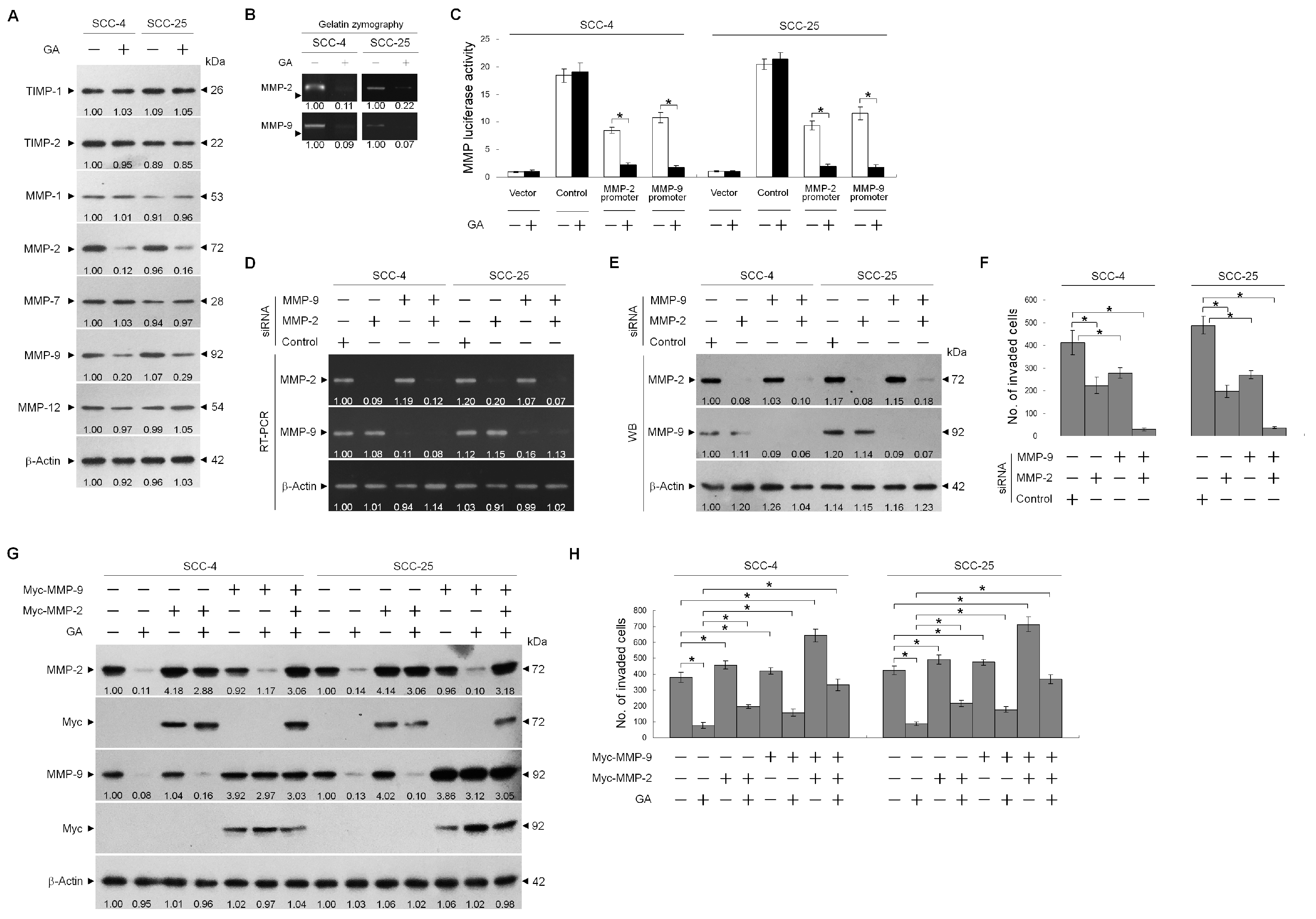



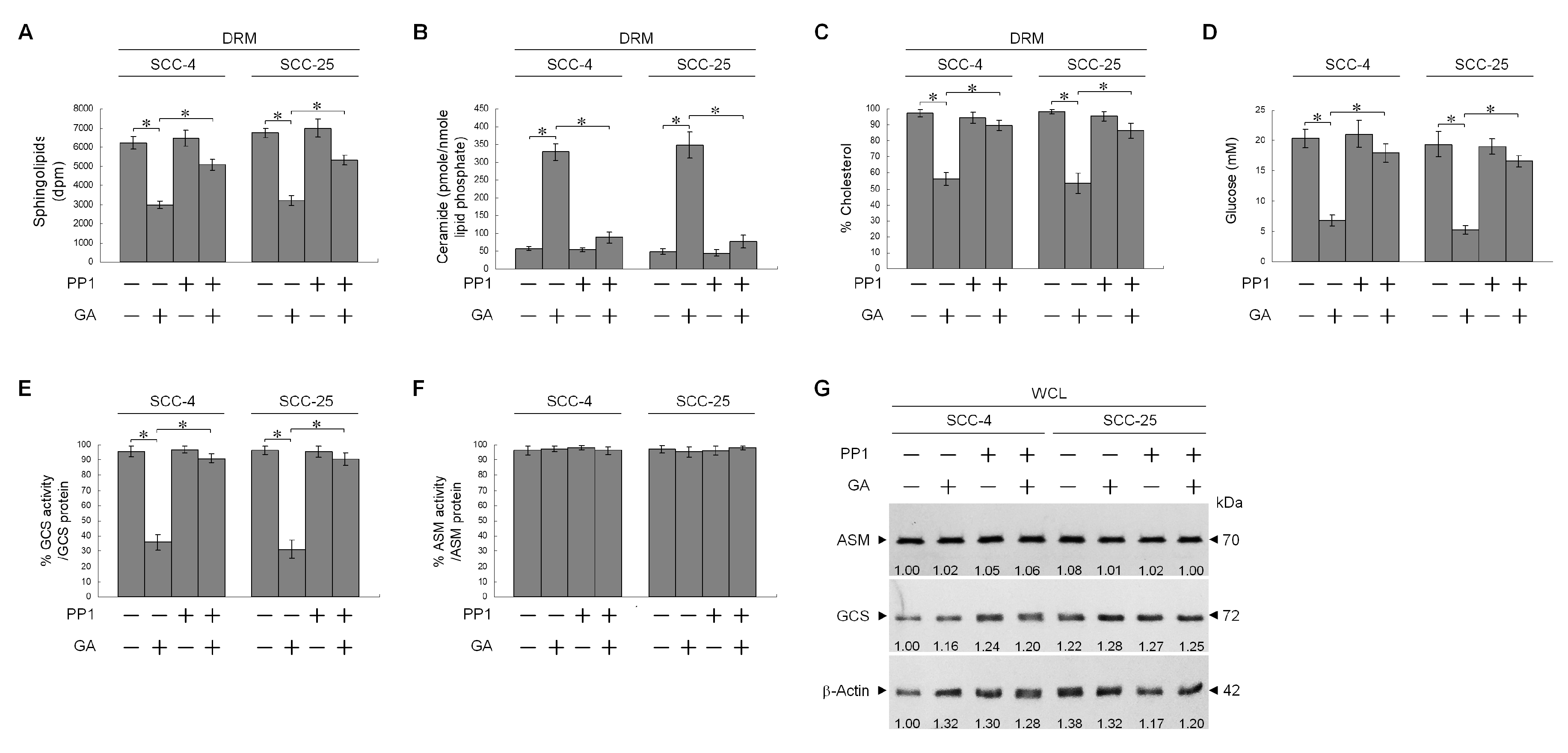
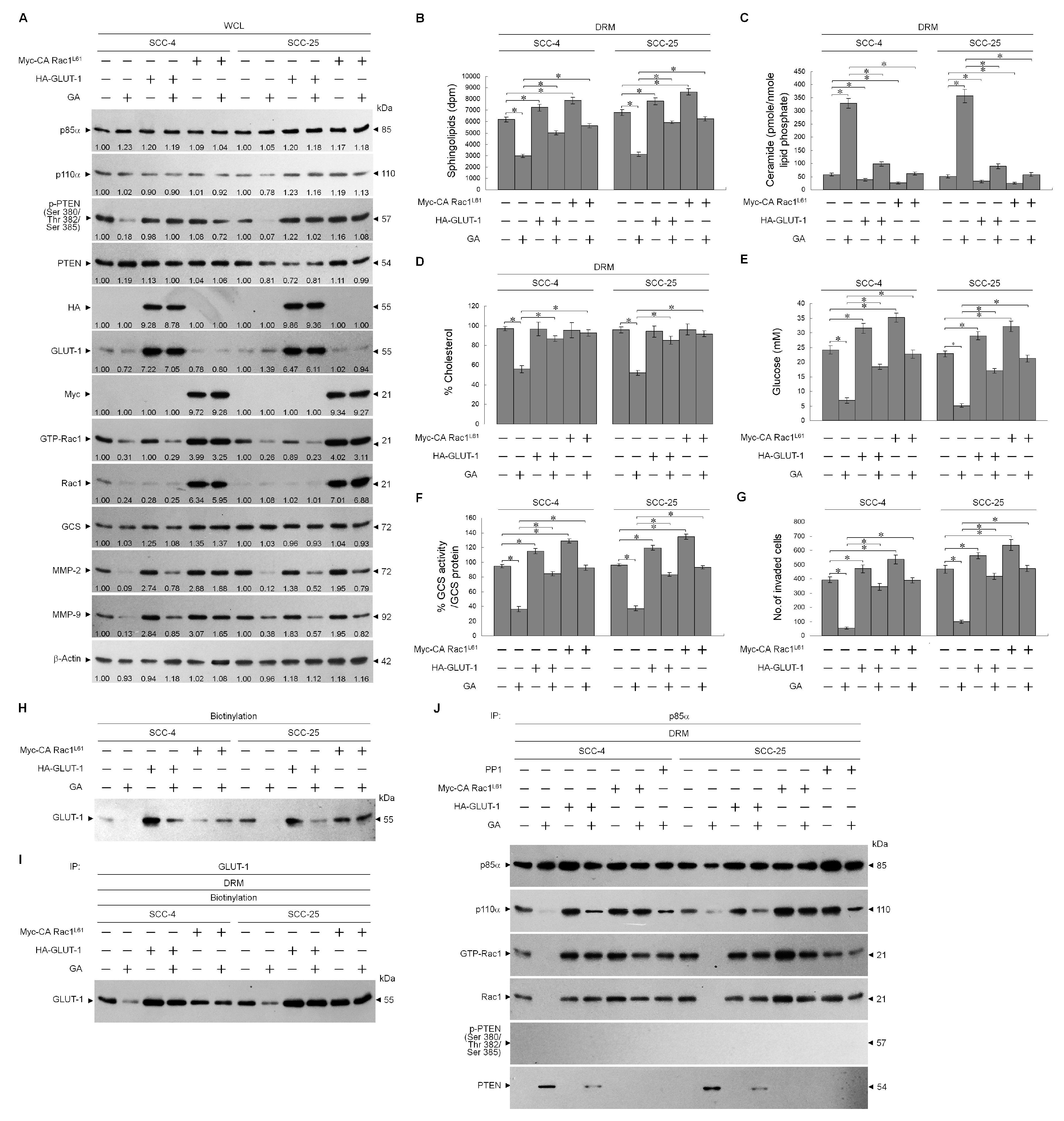
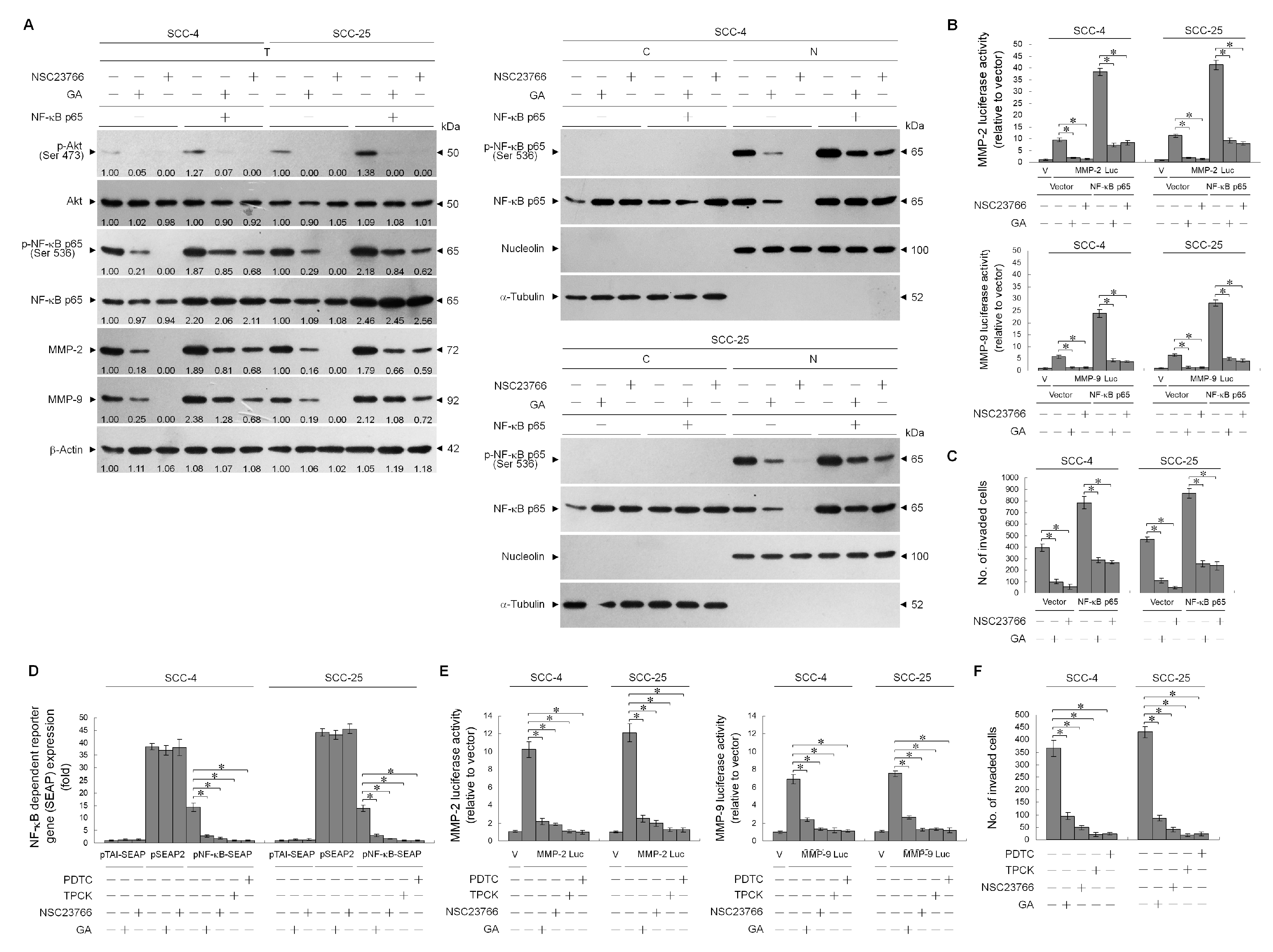

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.-W.; Wang, S.-G.; Lee, C.-H.; Chan, W.-L.; Lin, M.-L.; Chen, S.-S. Enforced C-Src Activation Causes Compartmental Dysregulation of PI3K and PTEN Molecules in Lipid Rafts of Tongue Squamous Carcinoma Cells by Attenuating Rac1-Akt-GLUT-1-Mediated Sphingolipid Synthesis. Int. J. Mol. Sci. 2020, 21, 5812. https://doi.org/10.3390/ijms21165812
Wu C-W, Wang S-G, Lee C-H, Chan W-L, Lin M-L, Chen S-S. Enforced C-Src Activation Causes Compartmental Dysregulation of PI3K and PTEN Molecules in Lipid Rafts of Tongue Squamous Carcinoma Cells by Attenuating Rac1-Akt-GLUT-1-Mediated Sphingolipid Synthesis. International Journal of Molecular Sciences. 2020; 21(16):5812. https://doi.org/10.3390/ijms21165812
Chicago/Turabian StyleWu, Chien-Wei, Shyang-Guang Wang, Ching-Hsiao Lee, Wen-Ling Chan, Meng-Liang Lin, and Shih-Shun Chen. 2020. "Enforced C-Src Activation Causes Compartmental Dysregulation of PI3K and PTEN Molecules in Lipid Rafts of Tongue Squamous Carcinoma Cells by Attenuating Rac1-Akt-GLUT-1-Mediated Sphingolipid Synthesis" International Journal of Molecular Sciences 21, no. 16: 5812. https://doi.org/10.3390/ijms21165812