Decreased Expression of Canstatin in Rat Model of Monocrotaline-Induced Pulmonary Arterial Hypertension: Protective Effect of Canstatin on Right Ventricular Remodeling

Abstract
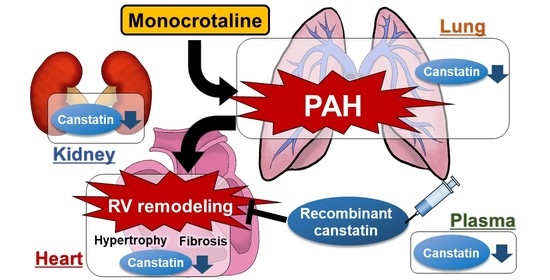
:
1. Introduction
2. Results
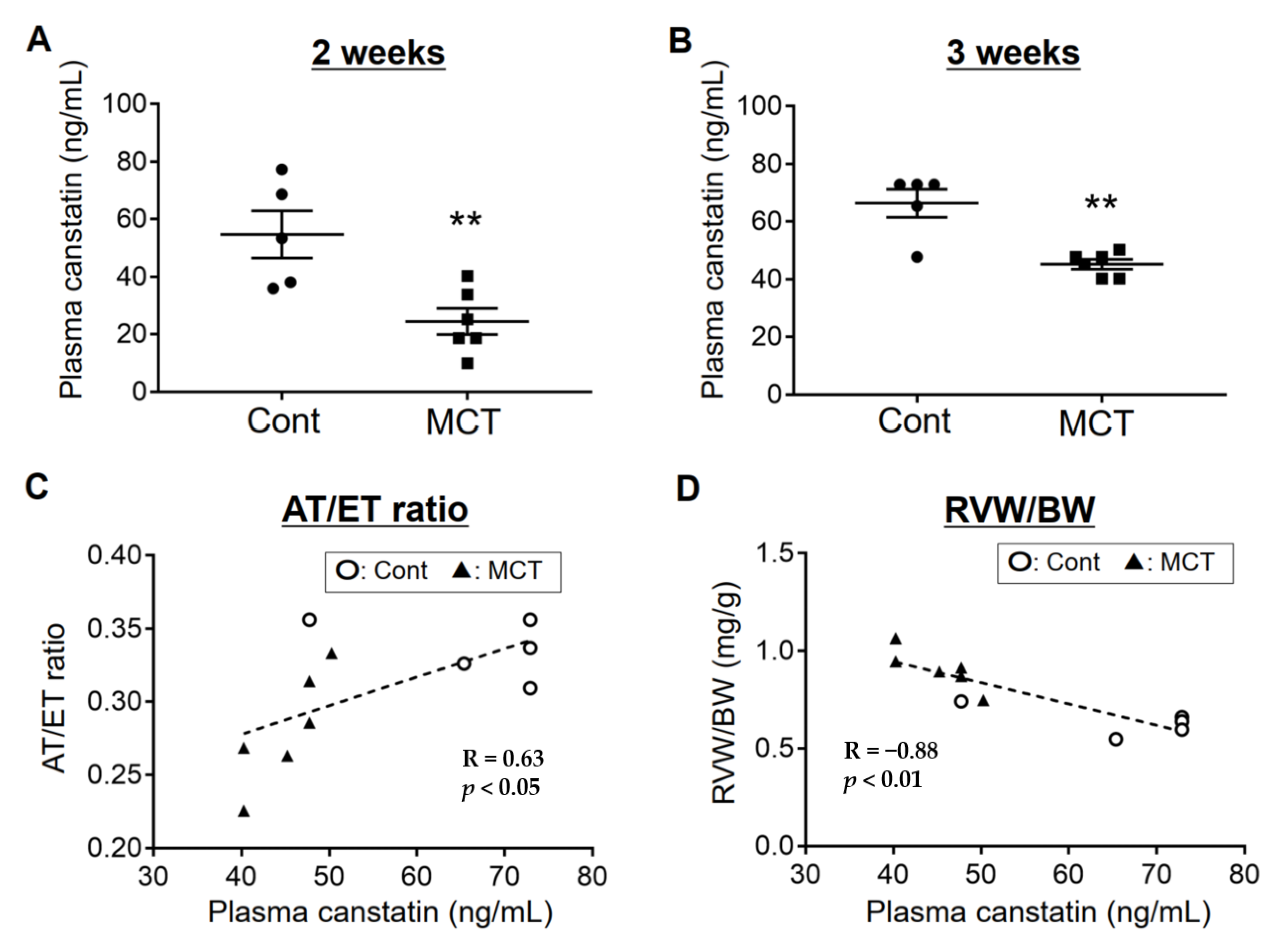
2.1. Plasma Concentration of Canstatin Was Decreased in PAH Rats, Which Was Correlated with Pathological Conditions
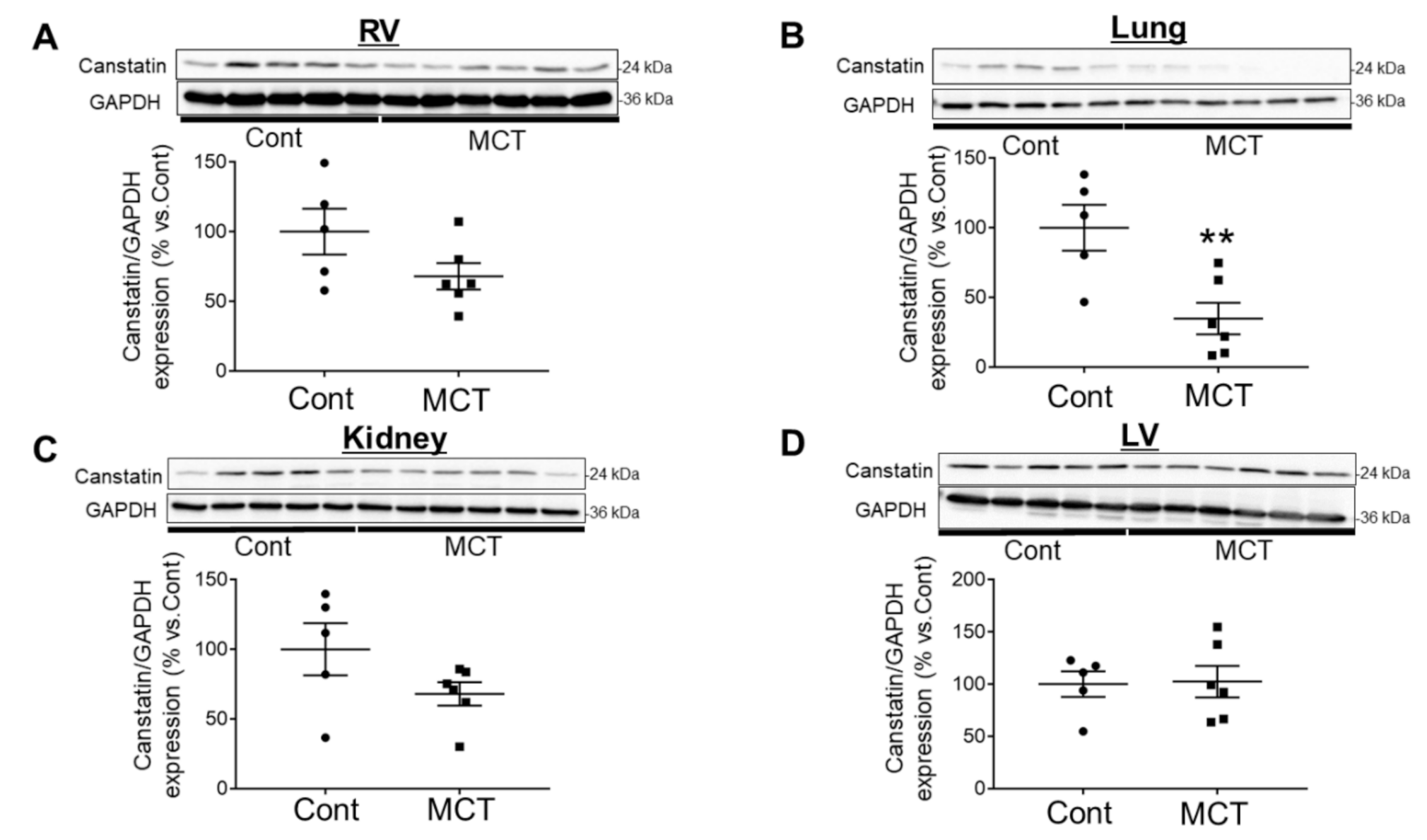
2.2. Expression of Canstatin in RV, Lung and Kidney Was Decreased in PAH Rats
2.3. Canstatin Had No Effect on Monocrotaline-Induced PAH
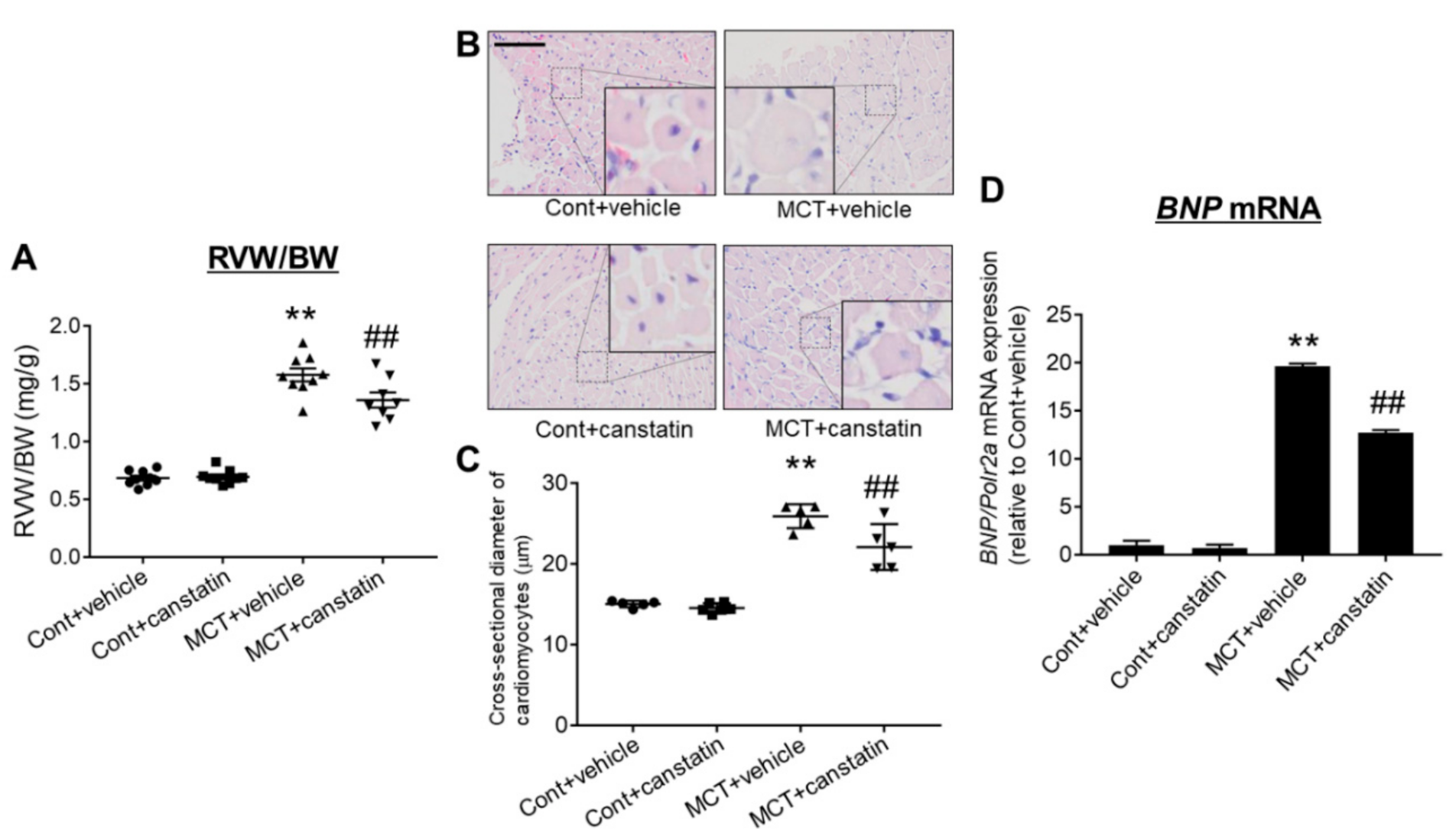
2.4. Canstatin Improved Monocrotaline-Induced RV Hypertrophy
2.5. Canstatin Improved Monocrotaline-Induced RV Fibrosis
3. Discussion
4. Materials and Methods
4.1. Regents and Antibodies
4.2. Animal Study
4.3. Measurement of Plasma Canstatin by Sandwich ELISA
4.4. Echocardiography
4.5. Western Blotting
4.6. Measurement of PA Pressure
4.7. Histological Analysis
4.8. qRT-PCR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PAH | pulmonary arterial hypertension |
| RV | right ventricle |
| BNP | brain natriuretic peptide |
| ELISA | enzyme-linked immunosorbent assay |
| MCT | monocrotaline-injected rats |
| Cont | saline-injected rats |
| AT | acceleration time |
| ET | ejection time |
| LW | lung weight |
| RVW | RV weight |
| BW | body weight |
| LV | left ventricle |
| LVW | LV weight |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| TAPSE | tricuspid annular plane systolic excursion |
| α-SMA | α-smooth muscle actin |
| TGF-β | transforming growth factor-β |
| NFAT | nuclear factor of activated T cells |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| EDTA | ethylenediaminetetraacetic acid |
| HE | hematoxylin and eosin |
| DAB | diaminobenzidine |
| Polr2a | RNA polymerase II subunit A |
| S.E.M. | standard error of the mean |
References
- Boucherat, O.; Vitry, G.; Trinh, I.; Paulin, R.; Provencher, S.; Bonnet, S. The cancer theory of pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Rain, S.; Handoko, M.L.; Vonk Noordegraaf, A.; Bogaard, H.J.; Van Der Velden, J.; De Man, F.S. Pressure-overload-induced right heart failure. Pflügers Arch. Eur. J. Physiol. 2014, 466, 1055–1063. [Google Scholar] [CrossRef]
- van de Veerdonk, M.C.; Bogaard, H.J.; Voelkel, N.F. The right ventricle and pulmonary hypertension. Heart Fail. Rev. 2016, 21, 259–271. [Google Scholar] [CrossRef]
- Pezzuto, B.; Badagliacca, R.; Poscia, R.; Ghio, S.; D’Alto, M.; Vitulo, P.; Mulè, M.; Albera, C.; Volterrani, M.; Fedele, F.; et al. Circulating biomarkers in pulmonary arterial hypertension: Update and future direction. J. Hear. Lung Transplant. 2015, 34, 282–305. [Google Scholar] [CrossRef]
- Kamphaus, G.D.; Colorado, P.C.; Panka, D.J.; Hopfer, H.; Ramchandran, R.; Torre, A.; Maeshima, Y.; Mier, J.W.; Sukhatme, V.P.; Kalluri, R. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J. Biol. Chem. 2000, 275, 1209–1215. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, A.; Mitsui, A.; Okada, M.; Yamawaki, H. Cathepsin S degrades arresten and canstatin in infarcted area after myocardial infarction in rats. J. Vet. Med. Sci. 2019, 81, 522–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, A.; Okada, M.; Yamawaki, H. Pathophysiological roles of canstatin on myofibroblasts after myocardial infarction in rats. Eur. J. Pharmacol. 2017, 807, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, A.; Okada, M.; Yamawaki, H. Canstatin suppresses isoproterenol-induced cardiac hypertrophy through inhibition of calcineurin/nuclear factor of activated T-cells pathway in rats. Eur. J. Pharmacol. 2020, 871, 172849. [Google Scholar] [CrossRef] [PubMed]
- Imoto, K.; Kumatani, S.; Okada, M.; Yamawaki, H. Endostatin is protective against monocrotaline-induced right heart disease through the inhibition of T-type Ca2+channel. Pflügers Arch. Eur. J. Physiol. 2016, 468, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Iwase, M.; Kanazawa, H.; Kawata, N.; Yoshimori, Y.; Hashimoto, K.; Yokoi, T.; Noda, A.; Takagi, K.; Koike, Y.; et al. Progressive development of pulmonary hypertension leading to right ventricular hypertrophy assessed by echocardiography in rats. Exp. Anim. 2003, 52, 285–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Lu, Z.; Zhang, Y.; Geng, S.; Xu, M.; Xu, L.; Huang, Y.; Zhuang, P.; Zhang, Y. Stage-dependent changes of β2-adrenergic receptor signaling in right ventricular remodeling in monocrotaline-induced pulmonary arterial hypertension. Int. J. Mol. Med. 2018, 41, 2493–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.; Binder, T. Echocardiographic evaluation of the right heart. Wien. Klin. Wochenschr. 2018, 130, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.J.; Chen, I.C.; Li, H.H.; Huang, C.C. EP4 agonist L-902,688 suppresses EndMT and attenuates right ventricular cardiac fibrosis in experimental pulmonary arterial hypertension. Int. J. Mol. Sci. 2018, 19, 727. [Google Scholar] [CrossRef] [Green Version]
- Schumann, C.; Lepper, P.M.; Frank, H.; Schneiderbauer, R.; Wibmer, T.; Kropf, C.; Stoiber, K.M.; Rdiger, S.; Kruska, L.; Krahn, T.; et al. Circulating biomarkers of tissue remodelling in pulmonary hypertension. Biomarkers 2010, 15, 523–532. [Google Scholar] [CrossRef]
- Damico, R.; Kolb, T.M.; Valera, L.; Wang, L.; Housten, T.; Tedford, R.J.; Kass, D.A.; Rafaels, N.; Gao, L.; Barnes, K.C.; et al. Serum endostatin is a genetically determined predictor of survival in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2015, 191, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.J.; Hsu, H.C.; Ho, W.J.; Chang, G.J.; Pang, J.H.S.; Chen, W.J.; Huang, C.C.; Lai, Y.J. Cathepsin S promotes the development of pulmonary arterial hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2019, 317, 1–13. [Google Scholar] [CrossRef]
- Naranjo, M.; Lo, K.B.; Mezue, K.; Rangaswami, J. Effects of pulmonary hypertension and right ventricular function in short and long-term kidney function. Curr. Cardiol. Rev. 2019, 15, 3–11. [Google Scholar] [CrossRef]
- Steubl, D.; Kumar, S.V.; Tato, M.; Mulay, S.R.; Larsson, A.; Lind, L.; Risérus, U.; Renders, L.; Heemann, U.; Carlsson, A.C.; et al. Circulating cathepsin-S levels correlate with GFR decline and sTNFR1 and sTNFR2 levels in mice and humans. Sci. Rep. 2017, 7, 43538. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Cheng, F.; Yu, W.; Rao, T.; Li, W.; Zhao, S.; Zhou, X.; Ning, J. Cathepsin S regulates renal fibrosis in mouse models of mild and severe hydronephrosis. Mol. Med. Rep. 2019, 20, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.P.; Li, Z.S.; Zhu, R.M.; Tu, Z.X.; Gao, J.; Pan, X.; Gong, Y.F.; Jin, J.; Man, X.H.; Wu, H.Y.; et al. Effects of recombinant human canstatin protein in the treatment of pancreatic cancer. World J. Gastroenterol. 2006, 12, 6652–6657. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arroyo, J.G.; Farkas, L.; Alhussaini, A.A.; Farkas, D.; Kraskauskas, D.; Voelkel, N.F.; Bogaard, H.J. The monocrotaline model of pulmonary hypertension in perspective. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, 363–369. [Google Scholar] [CrossRef]
- Falcón, D.; Galeano-Otero, I.; Calderón-Sánchez, E.; Del Toro, R.; Martín-Bórnez, M.; Rosado, J.A.; Hmadcha, A.; Smani, T. TRP channels: Current perspectives in the adverse cardiac remodeling. Front. Physiol. 2019, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Wang, Y.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Hill, J.A.; Olson, E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Investig. 2006, 116, 3114–3126. [Google Scholar] [CrossRef]
- Martínez-Martínez, S.; Lozano-Vidal, N.; López-Maderuelo, M.D.; Jiménez-Borreguero, L.J.; Armesilla, Á.L.; Redondo, J.M. Cardiomyocyte calcineurin is required for the onset and progression of cardiac hypertrophy and fibrosis in adult mice. FEBS J. 2019, 286, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Sado, Y.; Inoue, S.; Tomono, Y.; Omori, H. Lymphocytes from enlarged iliac lymph nodes as fusion partners for the production of monoclonal antibodies after a single tail base immunization attempt. Acta Histochem. Cytochem. 2006, 39, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Kameshima, S.; Kakuda, C.; Okamura, Y.; Kodama, T.; Okada, M.; Yamawaki, H. Visceral adipose tissue-derived serine protease inhibitor prevents the development of monocrotaline-induced pulmonary arterial hypertension in rats. Pflügers Arch. Eur. J. Physiol. 2017, 469, 1425–1432. [Google Scholar] [CrossRef]
- Otani, K.; Yokoya, M.; Kodama, T.; Hori, K.; Matsumoto, K.; Okada, M.; Yamawaki, H. Plasma exosomes regulate systemic blood pressure in rats. Biochem. Biophys. Res. Commun. 2018, 503, 776–783. [Google Scholar] [CrossRef]
- Sugiyama, A.; Hirano, Y.; Okada, M.; Yamawaki, H. Endostatin stimulates proliferation and migration of myofibroblasts isolated from myocardial infarction model rats. Int. J. Mol. Sci. 2018, 19, 741. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cont | MCT | |
|---|---|---|
| BW (g) | 339 ± 8 | 287 ± 4 ** |
| AT/ET ratio | 0.34 ± 0.01 | 0.28 ± 0.02 ** |
| LW (mg) | 1157 ± 36 | 1576 ± 64 ** |
| RVW (mg) | 217 ± 14 | 260 ± 13 ** |
| RVW/BW ratio (mg/g) | 0.64 ± 0.03 | 0.90 ± 0.04 ** |
| Cont + Vehicle | Cont + Canstatin | MCT + Vehicle | MCT + Canstatin | |
|---|---|---|---|---|
| BW (g) | 241 ± 4 | 240 ± 5 | 186 ± 4 ** | 195 ± 6 |
| AT/ET ratio | 0.38 ± 0.01 | 0.40 ± 0.01 | 0.22 ± 0.01 ** | 0.23 ± 0.01 |
| TAPSE | 0.37 ± 0.01 | 0.39 ± 0.02 | 0.12 ± 0.00 ** | 0.15 ± 0.01 |
| (LV + S)W (mg) | 608 ± 14 | 596 ± 9 | 524 ± 10 ** | 577 ± 18 # |
| RVW (mg) | 164 ± 4 | 167 ± 6 | 293 ± 11 ** | 263 ± 11 |
| RVW/BW ratio (mg/g) | 0.68 ± 0.02 | 0.69 ± 0.02 | 1.58 ± 0.06 ** | 1.36 ± 0.07 ## |
| RVW/(LV + S)W ratio (mg/mg) | 0.27 ± 0.01 | 0.28 ± 0.01 | 0.56 ± 0.02 ** | 0.46 ± 0.01 ## |
| LW (mg) | 1033 ± 18 | 1013 ± 19 | 1424 ± 58 ** | 1491 ± 74 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sugiyama, A.; Kaisho, M.; Okada, M.; Otani, K.; Yamawaki, H. Decreased Expression of Canstatin in Rat Model of Monocrotaline-Induced Pulmonary Arterial Hypertension: Protective Effect of Canstatin on Right Ventricular Remodeling. Int. J. Mol. Sci. 2020, 21, 6797. https://doi.org/10.3390/ijms21186797
Sugiyama A, Kaisho M, Okada M, Otani K, Yamawaki H. Decreased Expression of Canstatin in Rat Model of Monocrotaline-Induced Pulmonary Arterial Hypertension: Protective Effect of Canstatin on Right Ventricular Remodeling. International Journal of Molecular Sciences. 2020; 21(18):6797. https://doi.org/10.3390/ijms21186797
Chicago/Turabian StyleSugiyama, Akira, Maina Kaisho, Muneyoshi Okada, Kosuke Otani, and Hideyuki Yamawaki. 2020. "Decreased Expression of Canstatin in Rat Model of Monocrotaline-Induced Pulmonary Arterial Hypertension: Protective Effect of Canstatin on Right Ventricular Remodeling" International Journal of Molecular Sciences 21, no. 18: 6797. https://doi.org/10.3390/ijms21186797
APA StyleSugiyama, A., Kaisho, M., Okada, M., Otani, K., & Yamawaki, H. (2020). Decreased Expression of Canstatin in Rat Model of Monocrotaline-Induced Pulmonary Arterial Hypertension: Protective Effect of Canstatin on Right Ventricular Remodeling. International Journal of Molecular Sciences, 21(18), 6797. https://doi.org/10.3390/ijms21186797