Thermally Stable Nitrothiacalixarene Chromophores: Conformational Study and Aggregation Behavior

 ,
,  , , ,
, , ,
Abstract
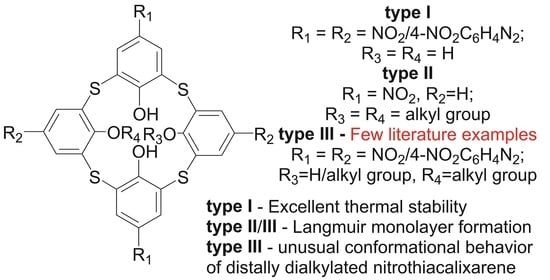
:
1. Introduction
2. Results
2.1. Design of the Chromophores
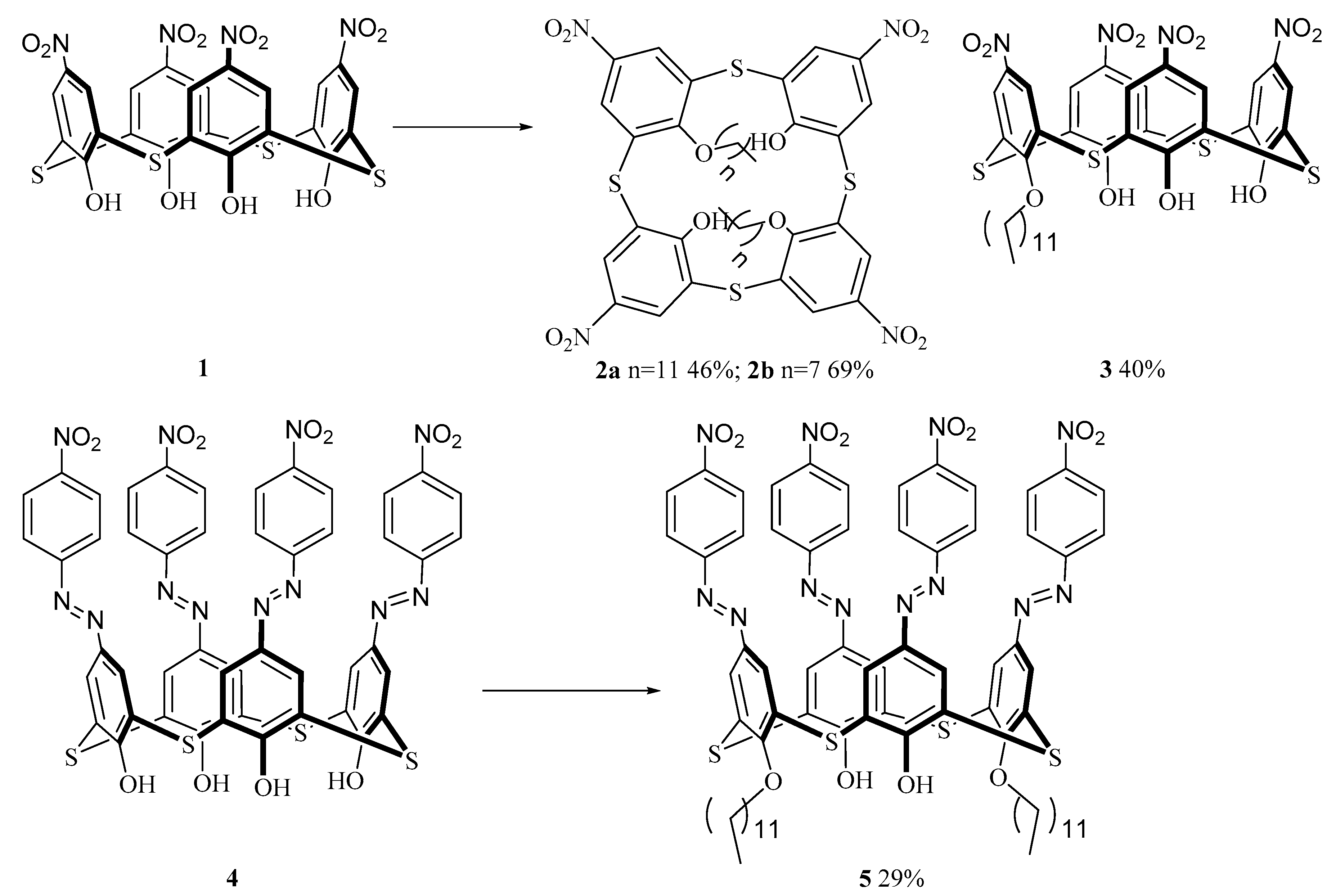
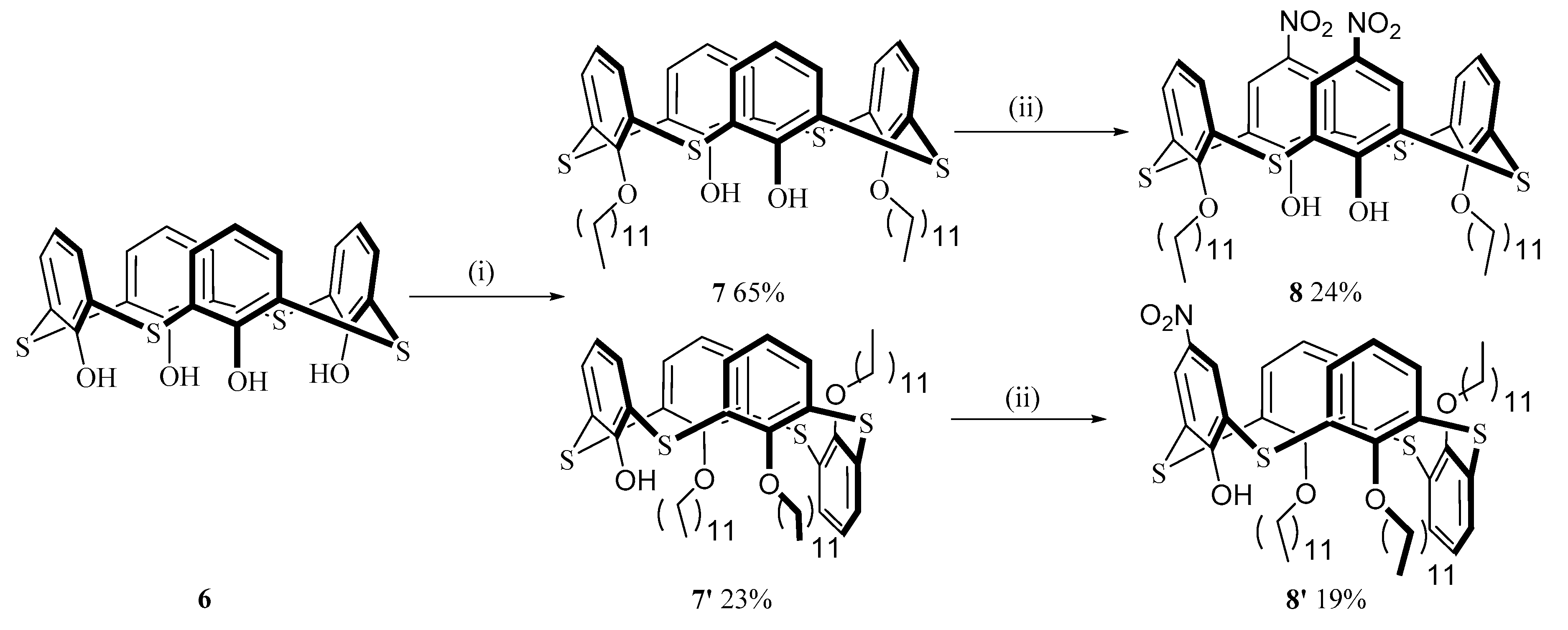
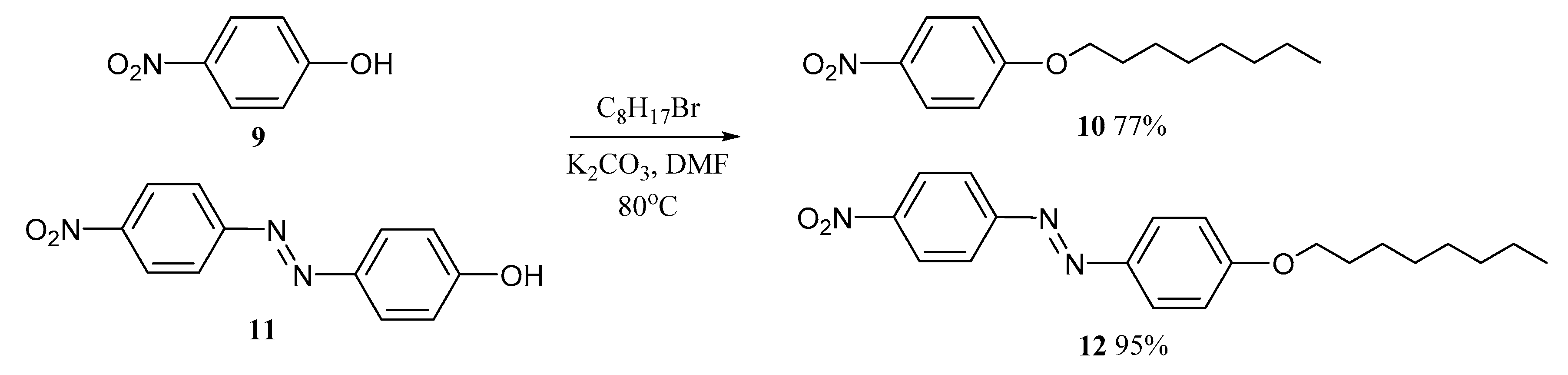
2.2. Synthesis of the Chromophores
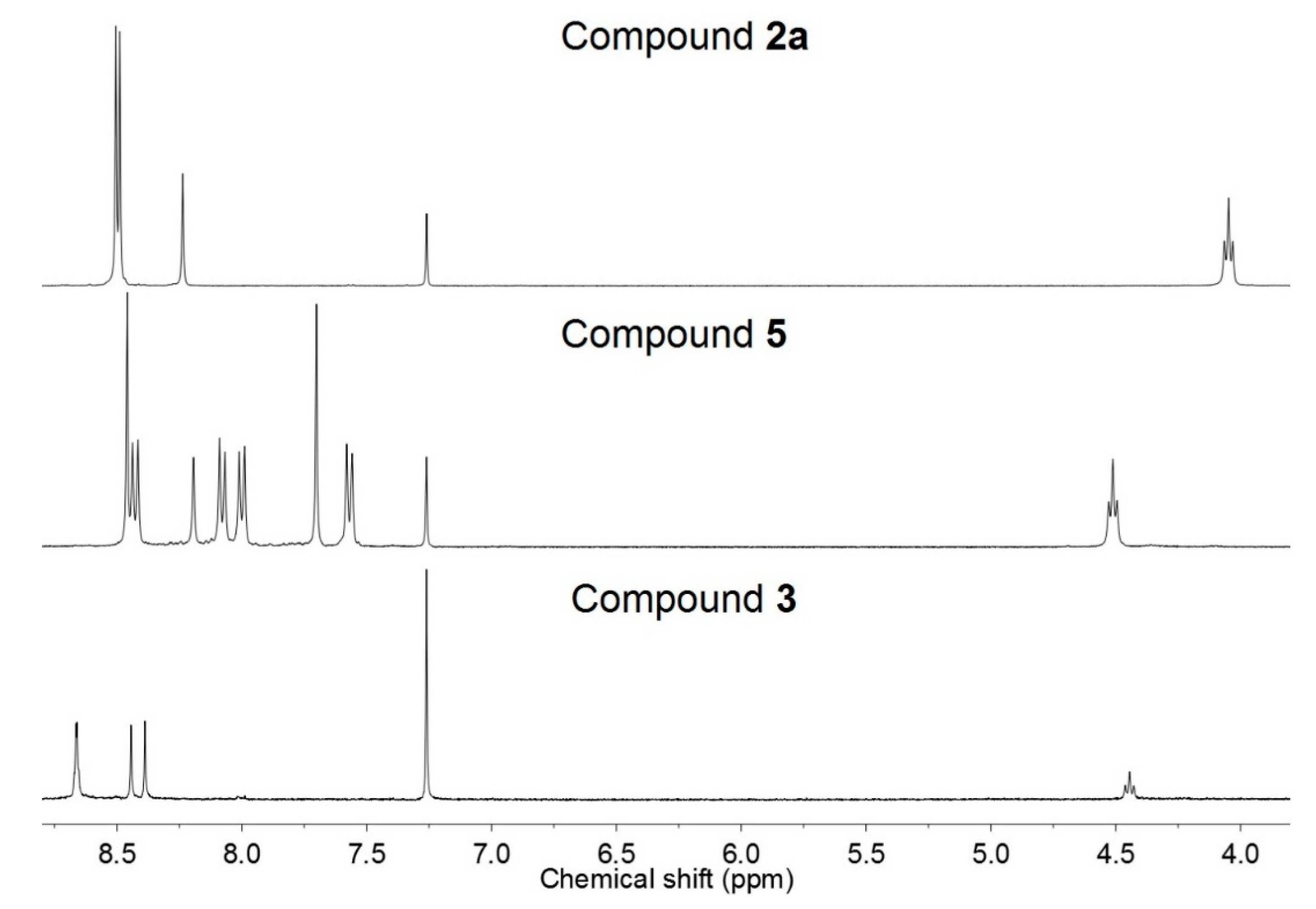
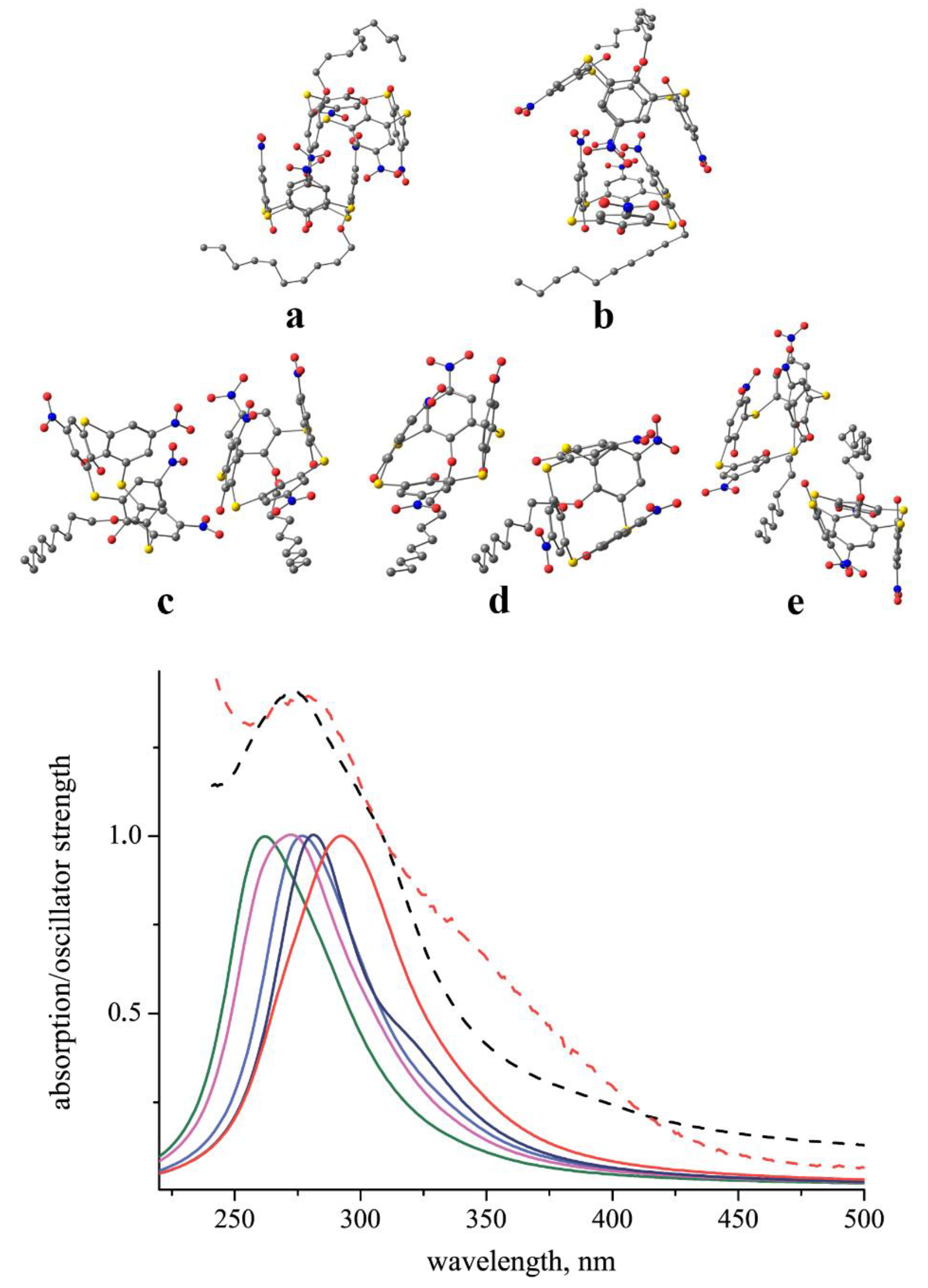
2.3. Conformational Study of the Chromophores
2.4. Thermal Stability Study
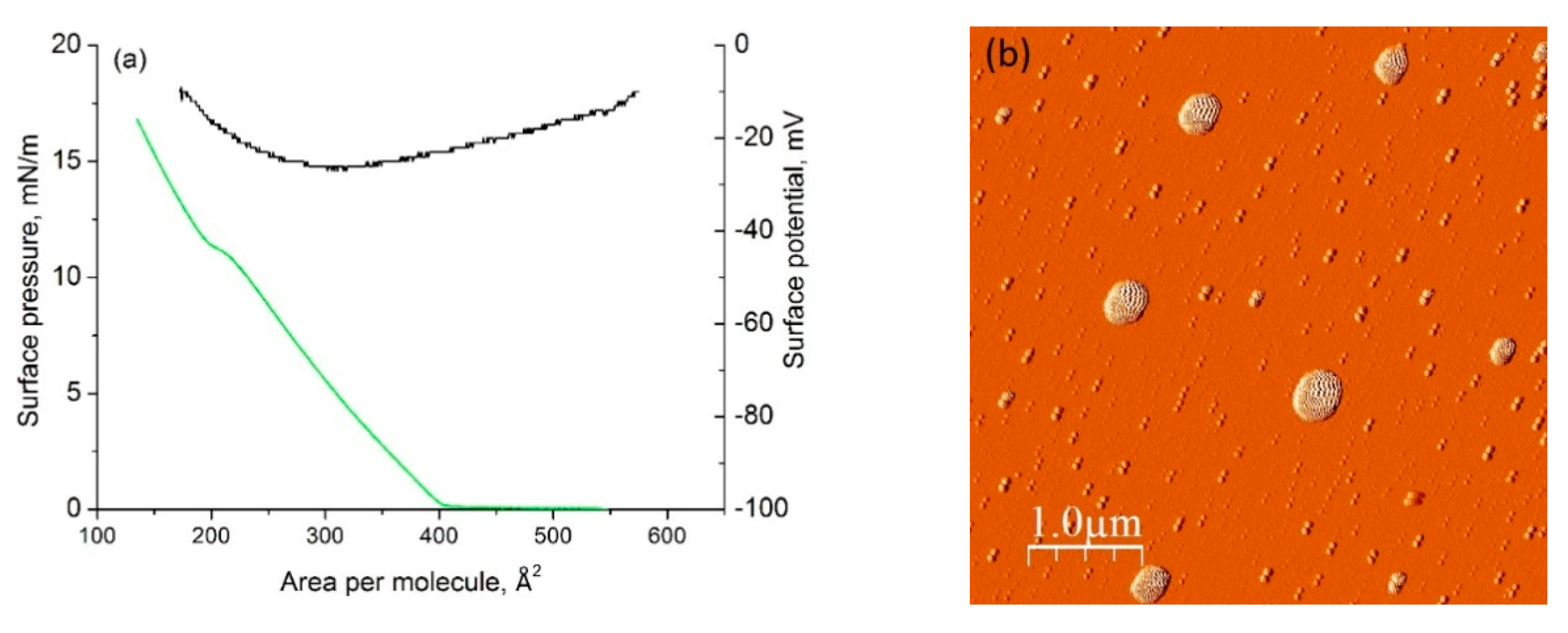
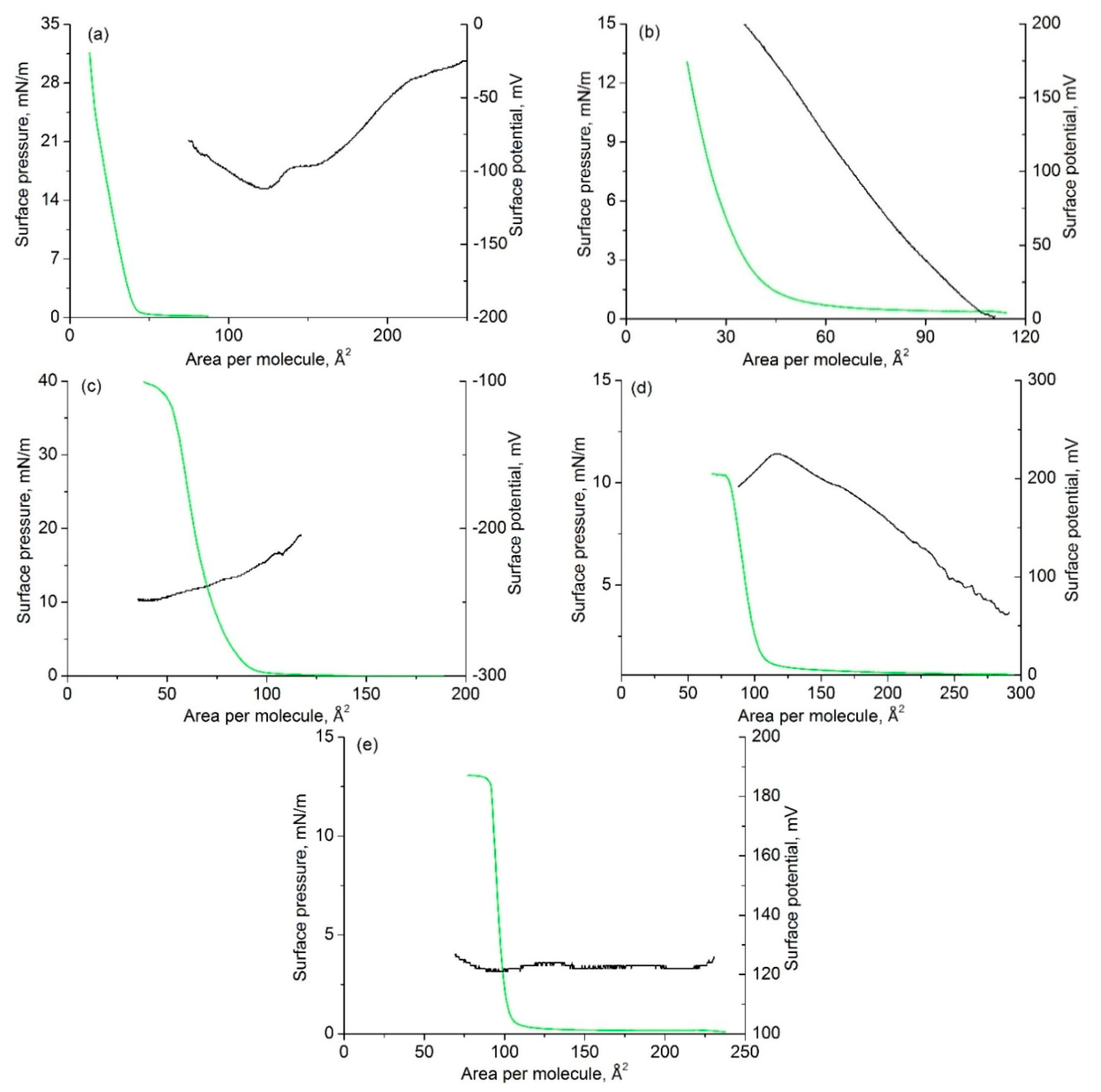
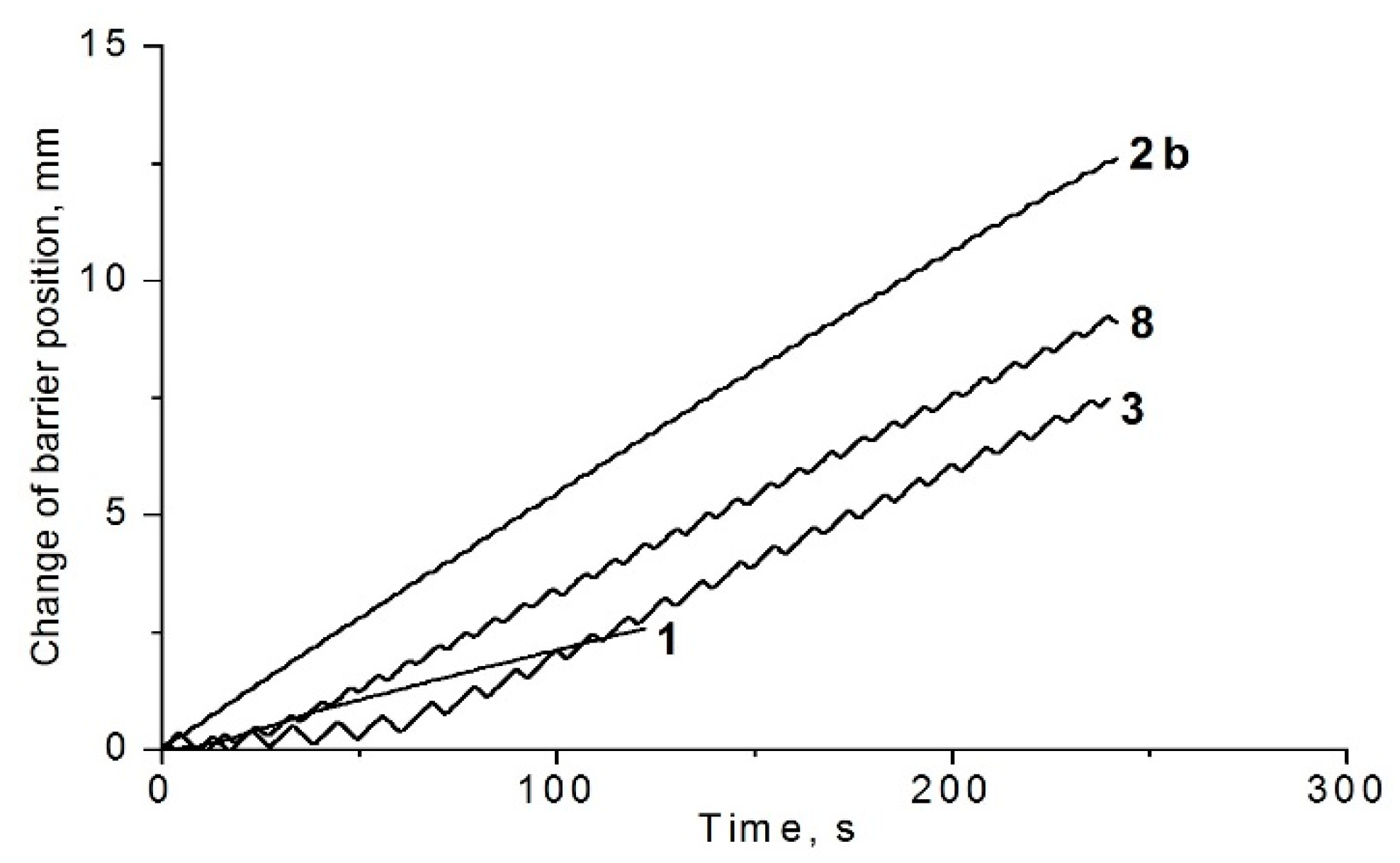
2.5. Langmuir Film Formation
2.6. Spectrophotometric Study of Nitro Chromophores in Solution, Air–Water Interface, and on Solid Substrate
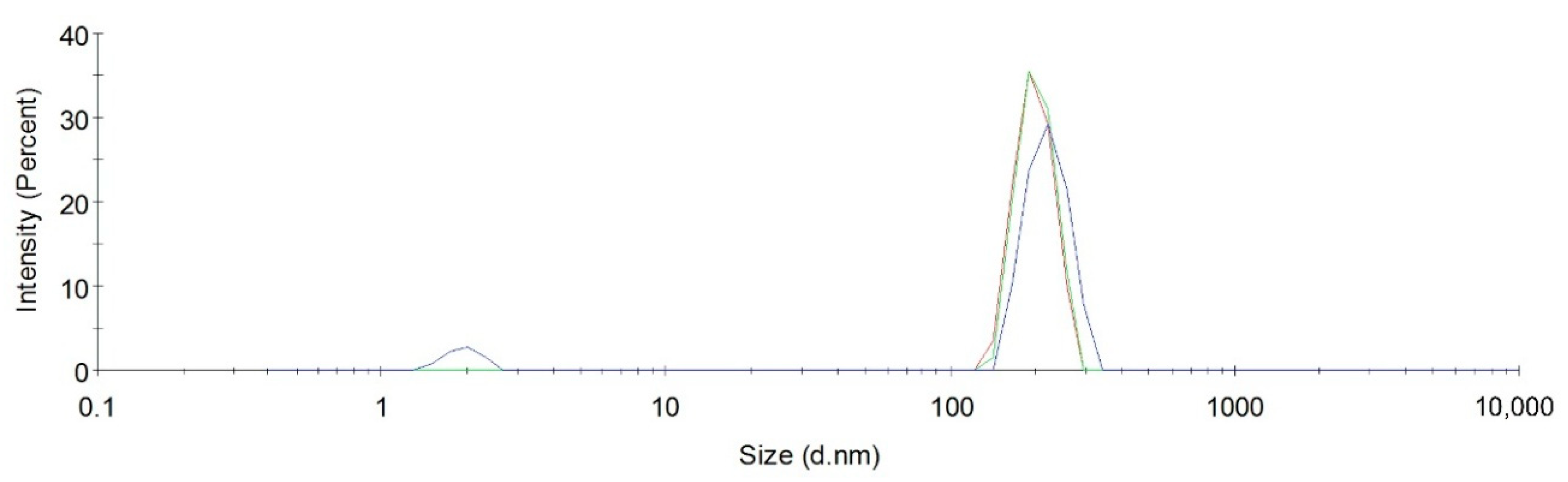
2.7. Contribution of Aggregation Phenomena to Optical Absorbance Characteristics of Nitrothiacalixarenes
3. Materials and Methods
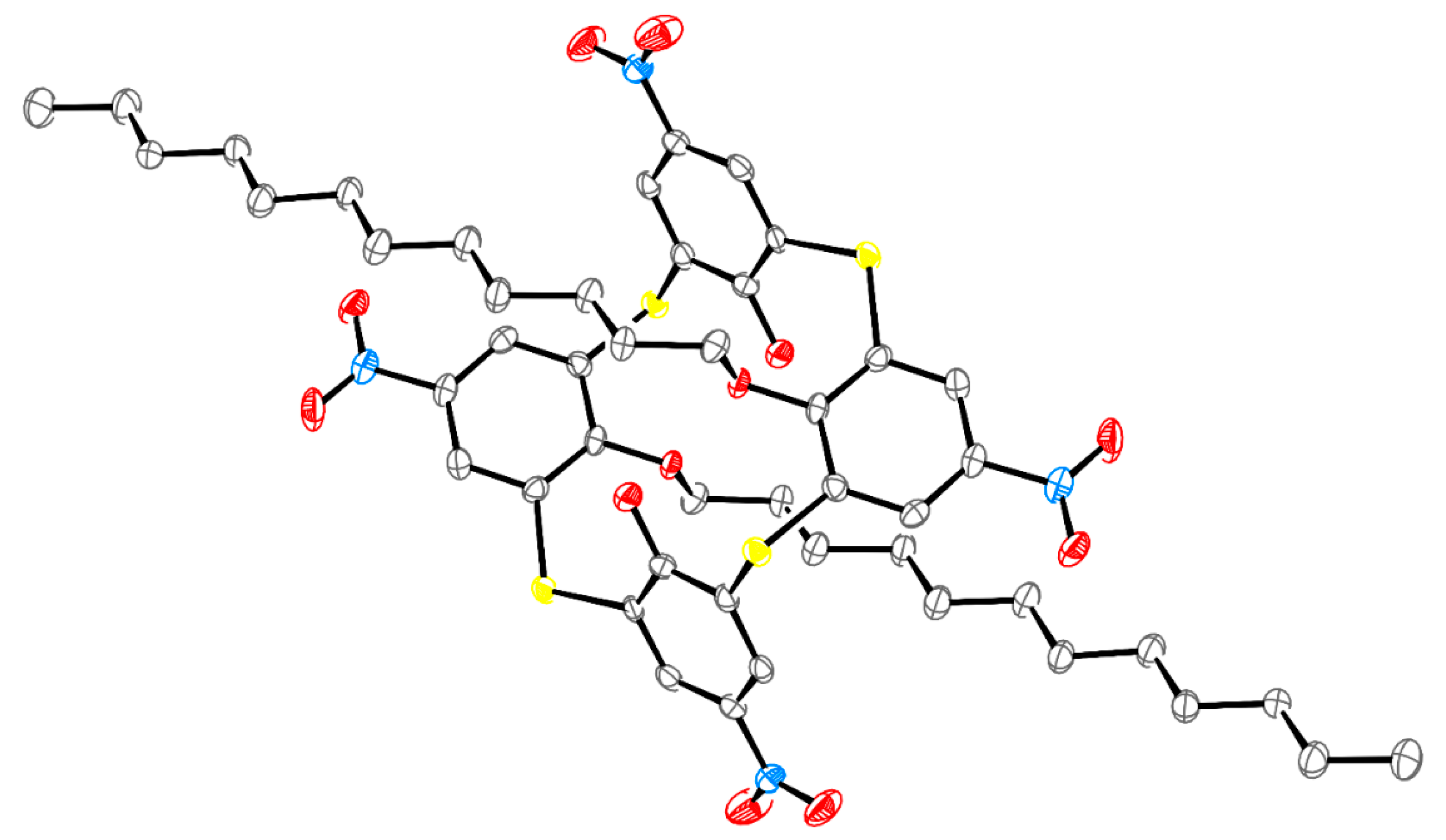
Crystallographic Data for 2a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NLO | Nonlinear optical |
| TPP | Triphenylphosphine |
| DEAD | Diethylazodicarboxylate |
| NMR | Nuclear magnetic resonance |
| CS | Chemical shift |
| DFT | Density functional theory |
| PC | Pinched cone |
| DC | Distorted cone |
| TG/DSC | Thermal gravimetry/differential scanning calorimetry |
| HNS | Hexanitrostilbene |
| PYX | 2,6-Bis(picrylamino)-3,5-dinitropyridine |
| LB | Langmuir–Blodgett |
| PSD | Particle size distribution |
| AFM | Atomic force microscopy |
| ITO | Indium-tin oxide |
| TR | Transfer ratio |
| UVRAS | Ultraviolet reflection-absorption spectroscopy |
| DLS | Dynamic light scattering |
| PXRD | Powder X-ray diffractometry |
| SHG | Second harmonic generation |
| IR | Infrared |
| MALDI | Matrix-assisted laser desorption–ionization |
| HRMS | High-resolution mass spectrometry |
| NA | Nitroaniline |
| TZVP | Valence triple-zeta polarization |
| B3LYP | Becke, 3-parameter, Lee–Yang–Parr |
References
- Würthner, F. Supermolecular Dye Chemistry; Springer: Berlin, Germany, 2005. [Google Scholar] [CrossRef]
- Davis, F.; Higson, S. Macrocycles: Construction, Chemistry and Nanotechnology Applications; Wiley: Chichester, UK, 2011. [Google Scholar] [CrossRef]
- Neri, P.; Sessler, J.L.; Wang, M.-X. Calixarenes and Beyond; Springer: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Fasting, C.; Schalley, C.A.; Weber, M.; Seitz, O.; Hecht, S.; Koksch, B.; Dernedde, J.; Graf, C.; Knapp, E.-W.; Haag, R. Multivalency as a chemical organization and action principle. Angew. Chem. Int. Ed. 2012, 51, 10472–10498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, H.-J. Strain effects determine the performance of artificial allosteric systems: Calixarenes as models. Chem. Commun. 2019, 55, 3433–3444. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-S. Calixarene-based supramolecular polymerization in solution. Chem. Soc. Rev. 2012, 41, 5907–5921. [Google Scholar] [CrossRef] [PubMed]
- Español, E.S.; Villamil, M.M. Calixarenes: Generalities and their role in improving the solubility, biocompatibility, stability, bioavailability, detection, and transport of biomolecules. Biomolecules 2019, 9, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreu, R.; Franco, S.; Garín, J.; Romero, J.; Villacampa, B.; Jesús Blesa, M.; Orduna, J. Multichromophoric calix[4]arenes: Effect of interchromophore distances on linear and nonlinear optical properties. ChemPhysChem 2012, 13, 3204–3209. [Google Scholar] [CrossRef] [PubMed]
- Liška, A.; Vojtíšek, P.; Fry, A.J.; Ludvík, J. Electrochemical and quantum chemical investigation of tetranitrocalix[4]arenes: Molecules with multiple redox centers. J. Org. Chem. 2013, 78, 10651–10656. [Google Scholar] [CrossRef] [PubMed]
- Thiampanya, P.; Muangsin, N.; Pulpoka, B. Azocalix[4]arene strapped calix[4]pyrrole: A confirmable fluoride sensor. Org. Lett. 2012, 14, 4050–4053. [Google Scholar] [CrossRef] [PubMed]
- Brake, M.; Böhmer, V.; Krämer, P.; Vogt, W.; Wortmann, R. O-Alkylated p-nitrocalix[4]arenes, synthesis, LB-monolayers and NLO-properties. Supramol. Chem. 1993, 2, 65–70. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, L.; Lu, G.-Y.; Zhang, C.-Z.; Jin, C.-M.; Liu, M. p-Nitrophenylazo calix[4]arenes, synthesis, monolayers and NLO-properties. Supramol. Chem. 2005, 17, 271–276. [Google Scholar] [CrossRef]
- Nishikubo, T.; Kameyama, A.; Kudo, H. Novel high performance materials. calixarene derivatives containing protective groups and polymerizable groups for photolithography, and calixarene derivatives containing active ester groups for thermal curing of epoxy resins. Polym. J. 2003, 35, 213–229. [Google Scholar] [CrossRef] [Green Version]
- Mačková, M.; Mikšátko, J.; Budka, J.; Eigner, V.; Cuřinova, P.; Lhoták, P. Chiral anion recognition by a ureido-thiacalix[4]arene ligand immobilized in the 1,3-alternate conformation. New J. Chem. 2015, 39, 1382–1389. [Google Scholar] [CrossRef]
- Klimentová, J.; Vojtíšek, P.; Sklenářová, M. Symmetrically tetrasubstituted p-nitrocalix[4]arenes: Synthesis, spectra and crystal structures. J. Mol. Struct. 2007, 871, 33–41. [Google Scholar] [CrossRef]
- Kasyan, O.; Rudzevich, V.; Bolte, M.; Böhmer, V. Tetraallyl ethers of thiacalix[4]arenes in the 1,3-alternate conformation. J. Chem. Cryst. 2011, 41, 332–337. [Google Scholar] [CrossRef]
- Ramanjaneyulu, P.S.; Singh, P.; Sayi, Y.S.; Chawla, H.M.; Ramakumar, K.L. Ion selective electrode for cesium based on 5-(4’-nitrophenylazo)25,27-bis(2-propyloxy)26,28-dihydroxycalix[4]arene. J. Hazard. Mater. 2010, 175, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- An, F.-J.; Xu, W.-Q.; Zheng, S.; Ma, S.-K.; Li, S.-Y.; Wang, R.-L.; Liu, J.-M. Bridging chiral calix[4]arenes: Description, optical resolution, and absolute configuration determination. Eur. J. Org. Chem. 2016, 2016, 1012–1016. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Chung, W.-S. Tetrazoles and para-substituted phenylazo-coupled calix[4]arenes as highly sensitive chromogenic sensors for Ca2+. Eur. J. Org. Chem. 2009, 2009, 4770–4776. [Google Scholar] [CrossRef]
- Kasyan, O.; Thondorf, I.; Bolte, M.; Kalchenko, V.; Böhmer, V. Unusual conformations of 1,3-dialkoxythiacalix[4]arenes in the solid state. Acta Cryst. C 2006, 62, o289–o294. [Google Scholar] [CrossRef]
- Oueslati, F.; Dumazet-Bonnamour, I.; Lamartine, R. New chromogenic azocalix[4]arene podands incorporating 2,2′-bipyridyl subunits. New J. Chem. 2003, 27, 644–647. [Google Scholar] [CrossRef]
- Oueslati, F.; Dumazet-Bonnamour, I.; Lamartine, R. Synthesis of new chromogenic 2,2′-bithiazoylcalix[4]arenes. Tetrahedron Lett. 2001, 42, 8177–8180. [Google Scholar] [CrossRef]
- Elçin, S.; Deligöz, H. A versatile approach toward chemosensor for Hg2+ based on para-substituted phenylazocalix[4]arene containing mono ethyl ester unit. Dye. Pigments 2014, 107, 166–173. [Google Scholar] [CrossRef]
- Mačková, M.; Himl, M.; Budka, J.; Pojarová, M.; Císařová, I.; Eigner, V.; Cuřínová, P.; Dvořáková, H.; Lhoták, P. Self-assembly of 5,11,17,23-tetranitro-25,26,27,28-tetramethoxythiacalix[4]arene with neutral molecules and its use for anion recognition. Tetrahedron 2013, 69, 1397–1402. [Google Scholar] [CrossRef]
- Lhoták, P.; Morávek, J.; Stibor, I. Diazo coupling: An alternative method for the upper rim amination of thiacalix[4]arenes. Tetrahedron Lett. 2002, 43, 3665–3668. [Google Scholar] [CrossRef]
- Muravev, A.A.; Laishevtsev, A.I.; Galieva, F.B.; Bazanova, O.B.; Rizvanov, I.K.; Korany, A.; Solovieva, S.E.; Antipin, I.S.; Konovalov, A.I. Azide-akyne click approach to the preparation of dendrimer-type multi(thia)calix[4]arenes with triazole linkers. Macroheterocycles 2017, 10, 203–214. [Google Scholar] [CrossRef]
- Muravev, A.A.; Solovieva, S.E.; Kochetkov, E.N.; Mel’nikova, N.B.; Safiullin, R.A.; Kadirov, M.K.; Latypov, S.K.; Antipin, I.S.; Konovalov, A.I. Thiacalix[4]monocrowns substituted by sulfur-containing anchoring groups: New ligands for gold surface modification. Macroheterocycles 2013, 6, 302–307. [Google Scholar] [CrossRef]
- Muravev, A.A.; Knyazeva, M.V.; Safiullin, R.A.; Shokurov, A.V.; Solovieva, S.E.; Selektor, S.L.; Antipin, I.S.; Konovalov, A.I. Nitrothiacalixarenes with alkyl groups at the lower rim: Design, synthesis and aggregation behaviour at the air–water interface and in solution. Mendeleev Commun. 2017, 27, 413–415. [Google Scholar] [CrossRef]
- Muravev, A.A.; Galieva, F.B.; Strel’nik, A.G.; Nugmanov, R.I.; Gruner, M.; Solov’eva, S.E.; Latypov, S.K.; Antipin, I.S.; Konovalov, A.I. Synthesis and structure of lower rim-substituted alkynyl derivatives of thiacalix[4]arene. Russ. J. Org. Chem. 2015, 51, 1334–1342. [Google Scholar] [CrossRef]
- Dvořáková, H.; Lang, J.; Vlach, J.; Sýkora, J.; Čajan, M.; Himl, M.; Pojarová, M.; Stibor, I.; Lhoták, P. Partially O-alkylated thiacalix[4]arenes: synthesis, molecular and crystal structures, conformational behavior. J. Org. Chem. 2007, 72, 7157–7166. [Google Scholar] [CrossRef]
- Desroches, C.; Parola, S.; Vocanson, F.; Perrin, M.; Lamartine, R.; Létoffé, J.-M.; Bouix, J. Nitration of thiacalix[4]arene using nitrosium nitrate complexes: Synthesis and characterization of tetranitro-, tetraamino-, and tetra(4-pyridylimino)tetrahydroxythiacalix[4]arene. New J. Chem. 2002, 26, 651–655. [Google Scholar] [CrossRef]
- Desroches, C.; Parola, S.; Vocanson, F.; Ehlinger, N.; Miele, P.; Lamartine, R.; Bouix, J.; Eriksson, A.; Lindgren, M.; Lopes, C. Synthesis, characterization and optical power limiting behaviour of phenylazo- and 4-nitrophenylazo-tetrahydroxytetrathiacalix[4]arene. J. Mater. Chem. 2001, 11, 3014–3017. [Google Scholar] [CrossRef]
- Desroches, C.; Lopes, C.; Kessler, V.; Parola, S. Design and synthesis of multifunctional thiacalixarenes and related metal derivatives for the preparation of sol-gel hybrid materials with non-linear optical properties. Dalton Trans. 2003, 11, 2085–2092. [Google Scholar] [CrossRef]
- Zhang, X.-C.; Xiong, H.-L.; Yang, H.-W.; Cheng, B. 14,16,34,36,54,56,74,76-Octanitro-2,4,6,8-tetraoxa-1,3,5,7(1,3)-tetrabenzenacyclooctaphane and its derivatives: Thermally stable explosives with outstanding properties. New J. Chem. 2017, 41, 5764–5769. [Google Scholar] [CrossRef]
- Gaines, G.L.J. Insoluble Monolayers at Liquid-Gas. Interfaces; Wiley: New York, NY, USA, 1966. [Google Scholar]
- Hu, X.; Shi, H.; Shi, X.; Zhu, Z.; Sun, Q.; Li, Y.; Yang, H. Selective nitration of thiacalix[4]arene and an investigation of its acid–base properties with a chemometric method. Bull. Chem. Soc. Jpn. 2005, 78, 138–141. [Google Scholar] [CrossRef]
- Ye, Z.; Pang, S.; He, W.; Shi, X.; Guo, Z.; Zhu, L. Copper(II) ion induced monolayer formation of p-tert-butylthiacalix[4]arene at the air–water interface. Spectrochim. Acta Part. A 2001, 57, 1443–1447. [Google Scholar] [CrossRef]
- Armarego, W.L.F. Purification of Laboratory Chemicals; Armarego, W.L.F., Chai, C.L.L., Eds.; Butterworth-Heinemann: Oxford, UK, 2009. [Google Scholar]
- Yang, P.-C.; Lu, Y.-L.; Li, C.-Y. Synthesis and characterization of photoactive azobenzene-based chromophores containing a bulky cholesteryl moiety. J. Mol. Struct. 2012, 1015, 129–137. [Google Scholar] [CrossRef]
- Akdas, H.; Bringel, L.; Graf, E.; Hosseini, M.W.; Mislin, G.; Pansanel, J.; De Cian, A.; Fischer, J. Thiacalixarenes: Synthesis and structural analysis of thiacalix[4]arene and of p-tert-butylthiacalix[4]arene. Tetrahedron Lett. 1998, 39, 2311–2314. [Google Scholar] [CrossRef]
- Stuchebryukov, S.D.; Selektor, S.L.; Silantieva, D.A.; Shokurov, A.V. Nonradiative energy transfer in mixed Langmuir monolayers and Langmuir–Blodgett films of compounds of different chemical composition and structure. Prot. Met. Phys. Chem. Surf. 2013, 49, 189–197. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford CT, UK, 2016. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory. Chem. Phys. Lett. 1996, 256, 454–464. [Google Scholar] [CrossRef]
- Bauernschmitt, R.; Häser, M.; Treutler, O.; Ahlrichs, R. Calculation of excitation energies within time-dependent density functional theory using auxiliary basis set expansions. Chem. Phys. Lett. 1997, 264, 573–578. [Google Scholar] [CrossRef]
- Furche, F. On the density matrix based approach to time-dependent density functional response theory. J. Chem. Phys. 2001, 114, 5982–5992. [Google Scholar] [CrossRef]
- Rudberg, E.; Sałek, P. Calculations of two-photon charge-transfer excitations using Coulomb-attenuated density-functional theory. J. Chem. Phys. 2005, 123, 184108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, R.; Amos, R.D. The application of CAM-B3LYP to the charge-transfer band problem of the zincbacteriochlorin-bacteriochlorin complex. Chem. Phys. Lett. 2006, 420, 106–109. [Google Scholar] [CrossRef]
- Cai, Z.L.; Crossley, M.J.; Reimers, J.R.; Kobayashi, R.; Amos, R.D. Density functional theory for charge transfer: the nature of the N-Bands of porphyrins and chlorophylls revealed through CAM-B3LYP, CASPT2, and SAC-CI calculations. J. Phys. Chem. B 2006, 110, 15624–15632. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]

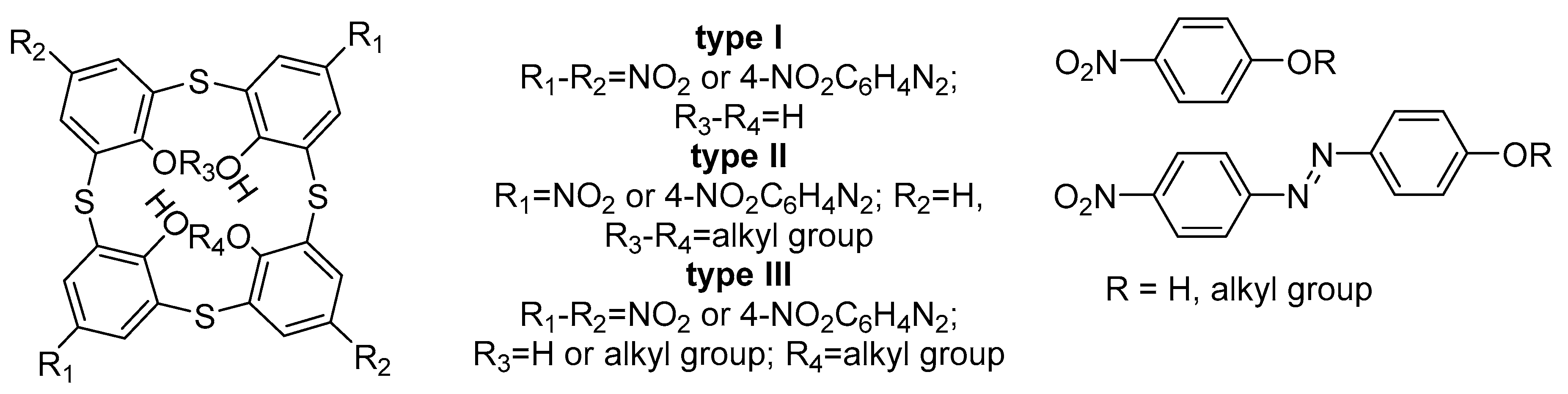



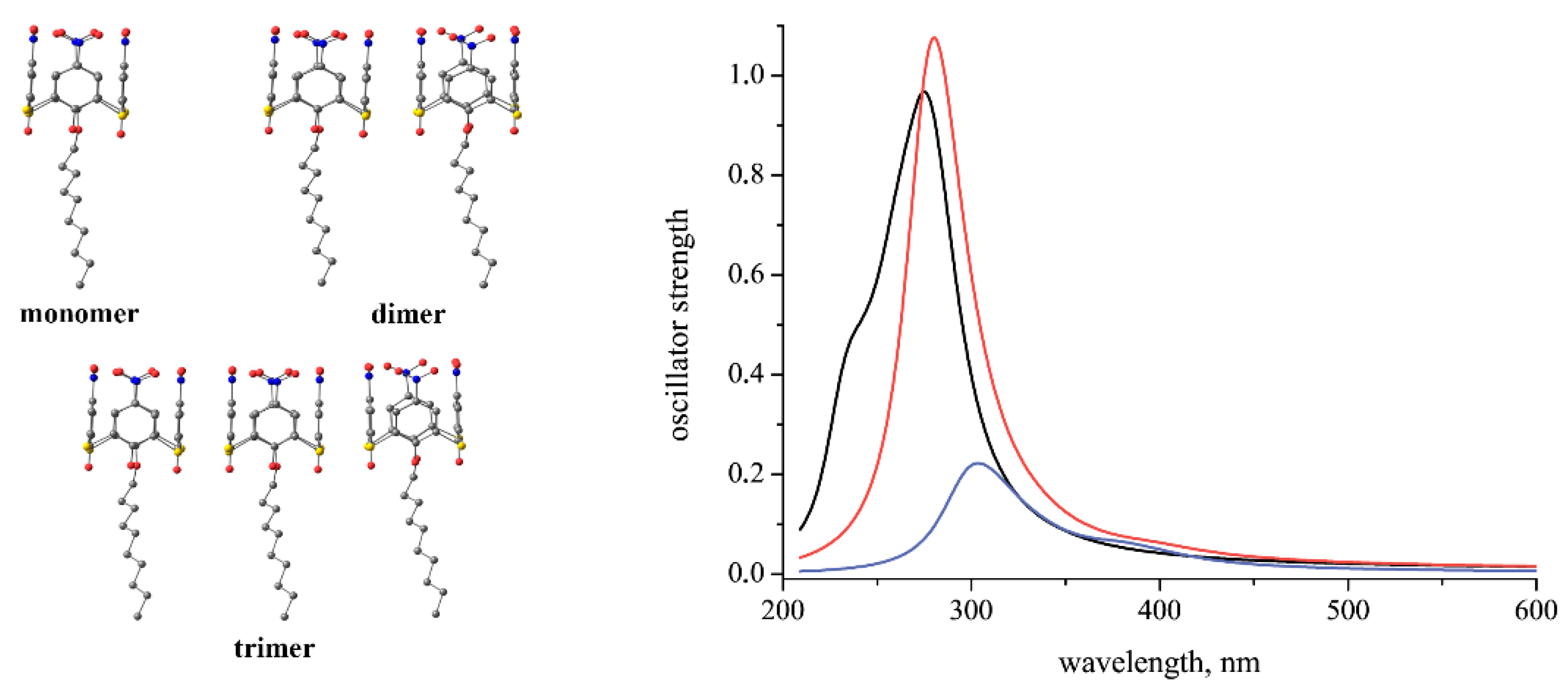

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2a | 2b | 3 | 4 | 5 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Td(TG),°C 1 | 363/360 | 278/266 | 283/253 | 243/218 | 340/291 | 295/282 | 311/299 | 193/181 | 226/210 | 280/262 | 295/269 |
| Td(DSC),°C 2 | 366 | 289 | 290 | 274 | 373 | 298 | 316 | 237 | 283 | 292 | 335 |
| Tm(DSC),°C 3 | 366 | 165 | 210 | 197 | 307 | 118 | 123 | 124 | n/a 4 | 204 | 85 |
| Time, h | Size, nm (Polydispersity Index) | |
|---|---|---|
| C2b = 10−4 M | C2b = 2 × 10−5 M | |
| Solvent—CHCl3 | ||
| 0 | 0.6, 10,100 (0.19) | 50 (0.22) |
| 1 | 10,100 (0.31) | 100 (0.25) |
| Solvent—MeOH/CHCl3 = 15:85 | ||
| 0 | 1 (0.29) | 1 (0.66) |
| 1 | 1 (0.31) | 1 (0.23) |
| Compound | A0, Å2/Molecule | πmax, mN/m | ΔV, mV, at 100 Å2 | |
|---|---|---|---|---|
| 1 | 45 | 30 | 36 | −100 |
| 2b | 400 | 12 | 27 | 10 |
| 3 | 90 | 40 | 120 | −230 |
| 4 | 45 | >13 | 20 | 0 |
| 5 | 105 | 10 | 8 | 200 |
| 8 | 107 | 13 | 144 | 120 |
| Compound | Roughness Rq, nm | Thickness h, nm | Transfer Ratio, TR |
|---|---|---|---|
| 8 | 2.7 (9.8) | 1.5 (12.0) | 1.0 (0.9–1.0) |
| 11 | 1.1 | 4.3 | 0.5 |
| 2b1 | 3.0 | 1.2 | 1.2 |
| 3 | 1.8 (4.0) | 4.3 (10.0) | 0.8 (0.8–0.9) |
| ITO | 2.63 | n/a | n/a |
| Compound | 8 | 1 | 2b | 3 | 5 | 4 |
|---|---|---|---|---|---|---|
| λmax, nm (CHCl3) | 277 | 273 | 274 | 273 | 354 | 368 |
| λmax, nm (air–water) | 283 | 317 | 292 | 285 | 365 | 380 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muravev, A.; Gerasimova, T.; Fayzullin, R.; Babaeva, O.; Rizvanov, I.; Khamatgalimov, A.; Kadirov, M.; Katsyuba, S.; Litvinov, I.; Latypov, S.; et al. Thermally Stable Nitrothiacalixarene Chromophores: Conformational Study and Aggregation Behavior. Int. J. Mol. Sci. 2020, 21, 6916. https://doi.org/10.3390/ijms21186916
Muravev A, Gerasimova T, Fayzullin R, Babaeva O, Rizvanov I, Khamatgalimov A, Kadirov M, Katsyuba S, Litvinov I, Latypov S, et al. Thermally Stable Nitrothiacalixarene Chromophores: Conformational Study and Aggregation Behavior. International Journal of Molecular Sciences. 2020; 21(18):6916. https://doi.org/10.3390/ijms21186916
Chicago/Turabian StyleMuravev, Anton, Tatiana Gerasimova, Robert Fayzullin, Olga Babaeva, Ildar Rizvanov, Ayrat Khamatgalimov, Marsil Kadirov, Sergey Katsyuba, Igor Litvinov, Shamil Latypov, and et al. 2020. "Thermally Stable Nitrothiacalixarene Chromophores: Conformational Study and Aggregation Behavior" International Journal of Molecular Sciences 21, no. 18: 6916. https://doi.org/10.3390/ijms21186916