Proteomic and Transcriptomic Patterns during Lipid Remodeling in Nannochloropsis gaditana

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Turbidostat Cultivation Dynamics, Lipids, and Fatty-Acids
2.2. Identification and Differential Expression of Proteins and Their Transcripts
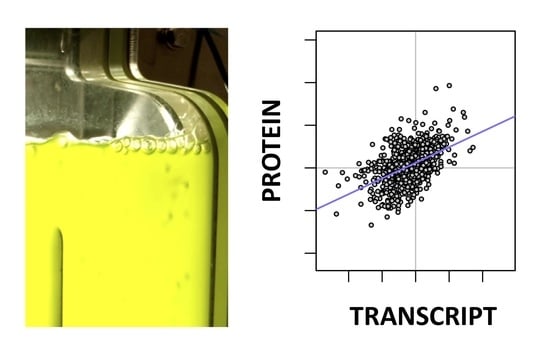
2.3. Correlation between the Nannochloropsis Proteome and Transcriptome
2.4. The Effect of Nitrogen and Phosphorus Stress on Subcellular Proteome Remodeling
2.5. Functional Enrichment Analysis of Differentially Expressed Proteins and Transcripts
2.6. Translation, Nitrogen Acquisition, and Metabolism
2.7. Tricarboxylic Acid (TCA) Cycle, Glycolytic Processes, and Oxidative Phosphorylation
2.8. Fatty-Acid and Acyl-CoA Metabolism
2.9. Polyunsaturated Fatty Acid (PUFA) Metabolism
2.10. Proteins Associated with TAG Biosynthesis and Storage in Oil Bodies
2.11. Glycerolipid and Phospholipid Biosynthesis
2.12. Lipase Activity and Lipid Catabolism
2.13. Polyketide Synthase, Fatty Acid Synthase, and Lipoxygenase Expression
3. Discussion
3.1. Global Correlation of Nannochloropsis Protein and Transcript Expression
3.2. N and P Deprivation Remodels Organelle Proteomes and Energy Metabolism
3.3. Lipid Metabolism and Remodeling
4. Materials and Methods
4.1. Cultivation
4.2. Sample Collection
4.3. Lipid Analysis
4.4. Nutrient Analysis
4.5. Proteomics
4.6. Transcriptomics
4.7. Gene Ontology and KEGG Pathway Gene Set Enrichment Analysis
4.8. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Quigg, A.; Finkel, Z.V.; Irwin, A.J.; Rosenthal, Y.; Ho, T.-Y.; Reinfelder, J.R.; Schofield, O.; Morel, F.M.; Falkowski, P.G. The evolutionary inheritance of elemental stoichiometry in marine phytoplankton. Nature 2003, 425, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Redfield, A.C. The influence of organisms on the composition of seawater. In The Sea; Wiley Interscience: New York, NY, USA, 1963; Volume 2, pp. 26–77. [Google Scholar]
- Browning, T.J.; Achterberg, E.P.; Rapp, I.; Engel, A.; Bertrand, E.M.; Tagliabue, A.; Moore, C.M. Nutrient co-limitation at the boundary of an oceanic gyre. Nature 2017, 551, 242. [Google Scholar] [CrossRef]
- Marañón, E.; Lorenzo, M.P.; Cermeño, P.; Mouriño-Carballido, B. Nutrient limitation suppresses the temperature dependence of phytoplankton metabolic rates. ISME J. 2018, 12, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Sommerfeld, M.; Jarvis, E.; Ghirardi, M.; Posewitz, M.; Seibert, M.; Darzins, A. Microalgal triacylglycerols as feedstocks for biofuel production: Perspectives and advances. Plant J. 2008, 54, 621–639. [Google Scholar] [CrossRef]
- Roth, M.S.; Cokus, S.J.; Gallaher, S.D.; Walter, A.; Lopez, D.; Erickson, E.; Endelman, B.; Westcott, D.; Larabell, C.A.; Merchant, S.S. Chromosome-level genome assembly and transcriptome of the green alga Chromochloris zofingiensis illuminates astaxanthin production. Proc. Natl. Acad. Sci. USA 2017, 114, E4296–E4305. [Google Scholar] [CrossRef]
- Elser, J.; Fagan, W.; Kerkhoff, A.; Swenson, N.; Enquist, B. Biological stoichiometry of plant production: Metabolism, scaling and ecological response to global change. New Phytol. 2010, 186, 593–608. [Google Scholar] [CrossRef]
- Toseland, A.; Daines, S.J.; Clark, J.R.; Kirkham, A.; Strauss, J.; Uhlig, C.; Lenton, T.M.; Valentin, K.; Pearson, G.A.; Moulton, V. The impact of temperature on marine phytoplankton resource allocation and metabolism. Nat. Clim. Chang. 2013, 3, 979–984. [Google Scholar] [CrossRef]
- Van Mooy, B.A.; Fredricks, H.F.; Pedler, B.E.; Dyhrman, S.T.; Karl, D.M.; Koblížek, M.; Lomas, M.W.; Mincer, T.J.; Moore, L.R.; Moutin, T. Phytoplankton in the ocean use non-phosphorus lipids in response to phosphorus scarcity. Nature 2009, 458, 69–72. [Google Scholar] [CrossRef]
- Cañavate, J.P.; Armada, I.; Hachero-Cruzado, I. Interspecific variability in phosphorus-induced lipid remodelling among marine eukaryotic phytoplankton. New Phytol. 2017, 213, 700–713. [Google Scholar] [CrossRef]
- Mühlroth, A.; Winge, P.; El Assimi, A.; Jouhet, J.; Marechal, E.; Hohmann-Marriott, M.F.; Vadstein, O.; Bones, A.M. Mechanisms of phosphorus acquisition and lipid class remodelling under P limitation in a marine microalga. Plant Physiol. 2017, 175, 1543–1559. [Google Scholar] [CrossRef]
- Li, J.; Han, D.; Wang, D.; Ning, K.; Jia, J.; Wei, L.; Jing, X.; Huang, S.; Chen, J.; Li, Y. Choreography of transcriptomes and lipidomes of Nannochloropsis reveals the mechanisms of oil synthesis in microalgae. Plant Cell 2014, 26, 1645–1665. [Google Scholar] [CrossRef]
- Rismani-Yazdi, H.; Haznedaroglu, B.Z.; Hsin, C.; Peccia, J. Transcriptomic analysis of the oleaginous microalga Neochloris oleoabundans reveals metabolic insights into triacylglyceride accumulation. Biotechnol. Biofuels 2012, 5, 74. [Google Scholar] [CrossRef]
- Peng, X.; Qin, Z.; Zhang, G.; Guo, Y.; Huang, J. Integration of the proteome and transcriptome reveals multiple levels of gene regulation in the rice dl2 mutant. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef]
- Li, J.J.; Chew, G.-L.; Biggin, M.D. Quantitating translational control: mRNA abundance-dependent and independent contributions and the mRNA sequences that specify them. Nucleic Acids Res. 2017, 45, 11821–11836. [Google Scholar] [CrossRef]
- De Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Molecular BioSystems 2009, 5, 1512–1526. [Google Scholar] [CrossRef]
- Vogel, C.; de Sousa Abreu, R.; Ko, D.; Le, S.Y.; Shapiro, B.A.; Burns, S.C.; Sandhu, D.; Boutz, D.R.; Marcotte, E.M.; Penalva, L.O. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol. Syst. Biol. 2010, 6, 400. [Google Scholar] [CrossRef]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef]
- Lahtvee, P.-J.; Sánchez, B.J.; Smialowska, A.; Kasvandik, S.; Elsemman, I.E.; Gatto, F.; Nielsen, J. Absolute Quantification of Protein and mRNA Abundances Demonstrate Variability in Gene-Specific Translation Efficiency in Yeast. Cell Syst. 2017, 4, 495–504. [Google Scholar] [CrossRef]
- Radakovits, R.; Jinkerson, R.E.; Fuerstenberg, S.I.; Tae, H.; Settlage, R.E.; Boore, J.L.; Posewitz, M.C. Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropsis gaditana. Nat. Commun. 2012, 3, 686. [Google Scholar] [CrossRef]
- Carpinelli, E.C.; Telatin, A.; Vitulo, N.; Forcato, C.; D’Angelo, M.; Schiavon, R.; Vezzi, A.; Giacometti, G.M.; Morosinotto, T.; Valle, G. Chromosome scale genome assembly and transcriptome profiling of Nannochloropsis gaditana in nitrogen depletion. Mol. Plant 2014, 7, 323–335. [Google Scholar] [CrossRef]
- Zienkiewicz, K.; Zienkiewicz, A.; Poliner, E.; Du, Z.-Y.; Vollheyde, K.; Herrfurth, C.; Marmon, S.; Farré, E.M.; Feussner, I.; Benning, C. Nannochloropsis, a rich source of diacylglycerol acyltransferases for engineering of triacylglycerol content in different hosts. Biotechnol. Biofuels 2017, 10, 8. [Google Scholar] [CrossRef]
- Ma, X.-N.; Chen, T.-P.; Yang, B.; Liu, J.; Chen, F. Lipid production from Nannochloropsis. Mar. Drugs 2016, 14, 61. [Google Scholar] [CrossRef]
- Calder, P.C. Very long chain omega-3 (n-3) fatty acids and human health. Eur. J. Lipid Sci. Technol. 2014, 116, 1280–1300. [Google Scholar] [CrossRef]
- Hulatt, C.J.; Wijffels, R.H.; Bolla, S.; Kiron, V. Production of fatty acids and protein by Nannochloropsis in flat-plate photobioreactors. PLoS ONE 2017, 12, e0170440. [Google Scholar] [CrossRef]
- Nelson, C.J.; Millar, A.H. Protein turnover in plant biology. Nat. Plants 2015, 1, 15017. [Google Scholar] [CrossRef]
- Floor, S.N.; Doudna, J.A. Tunable protein synthesis by transcript isoforms in human cells. Elife 2016, 5, e10921. [Google Scholar] [CrossRef]
- Calixto, C.P.; Guo, W.; James, A.B.; Tzioutziou, N.A.; Entizne, J.C.; Panter, P.E.; Knight, H.; Nimmo, H.; Zhang, R.; Brown, J.W. Rapid and dynamic alternative splicing impacts the Arabidopsis cold response transcriptome. Plant Cell 2018, 30, 1424–1444. [Google Scholar] [CrossRef]
- Rastogi, A.; Maheswari, U.; Dorrell, R.G.; Vieira, F.R.J.; Maumus, F.; Kustka, A.; McCarthy, J.; Allen, A.E.; Kersey, P.; Bowler, C. Integrative analysis of large scale transcriptome data draws a comprehensive landscape of Phaeodactylum tricornutum genome and evolutionary origin of diatoms. Sci. Rep. 2018, 8, 4834. [Google Scholar] [CrossRef]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A. RNA velocity of single cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef]
- Airoldi, E.M.; Miller, D.; Athanasiadou, R.; Brandt, N.; Abdul-Rahman, F.; Neymotin, B.; Hashimoto, T.; Bahmani, T.; Gresham, D. Steady-state and dynamic gene expression programs in Saccharomyces cerevisiae in response to variation in environmental nitrogen. Mol. Biol. Cell 2016, 27, 1383–1396. [Google Scholar] [CrossRef]
- Sforza, E.; Calvaruso, C.; La Rocca, N.; Bertucco, A. Luxury uptake of phosphorus in Nannochloropsis salina: Effect of P concentration and light on P uptake in batch and continuous cultures. Biochem. Eng. J. 2018, 134, 69–79. [Google Scholar] [CrossRef]
- Dong, H.-P.; Williams, E.; Wang, D.-z.; Xie, Z.-X.; Hsia, R.-c.; Jenck, A.; Halden, R.; Li, J.; Chen, F.; Place, A.R. Responses of Nannochloropsis oceanica IMET1 to long-term nitrogen starvation and recovery. Plant Physiol. 2013, 162, 1110–1126. [Google Scholar] [CrossRef]
- Plaxton, W.C.; Tran, H.T. Metabolic adaptations of phosphate-starved plants. Plant Physiol. 2011, 156, 1006–1015. [Google Scholar] [CrossRef]
- Remmers, I.M.; D’Adamo, S.; Martens, D.E.; de Vos, R.C.; Mumm, R.; America, A.H.; Cordewener, J.H.; Bakker, L.V.; Peters, S.A.; Wijffels, R.H. Orchestration of transcriptome, proteome and metabolome in the diatom Phaeodactylum tricornutum during nitrogen limitation. Algal Res. 2018, 35, 33–49. [Google Scholar] [CrossRef]
- Nobusawa, T.; Hori, K.; Mori, H.; Kurokawa, K.; Ohta, H. Differently localized lysophosphatidic acid acyltransferases crucial for triacylglycerol biosynthesis in the oleaginous alga Nannochloropsis. Plant J. 2017, 90, 547–559. [Google Scholar] [CrossRef]
- La Russa, M.; Bogen, C.; Uhmeyer, A.; Doebbe, A.; Filippone, E.; Kruse, O.; Mussgnug, J.H. Functional analysis of three type-2 DGAT homologue genes for triacylglycerol production in the green microalga Chlamydomonas reinhardtii. J. Biotechnol. 2012, 162, 13–20. [Google Scholar] [CrossRef]
- Yoneda, K.; Yoshida, M.; Suzuki, I.; Watanabe, M.M. Identification of a major lipid droplet protein in a marine diatom Phaeodactylum tricornutum. Plant Cell Physiol. 2016, 57, 397–406. [Google Scholar] [CrossRef]
- Vieler, A.; Brubaker, S.B.; Vick, B.; Benning, C. A lipid droplet protein of Nannochloropsis with functions partially analogous to plant oleosins. Plant Physiol. 2012, 158, 1562–1569. [Google Scholar] [CrossRef]
- Alboresi, A.; Perin, G.; Vitulo, N.; Diretto, G.; Block, M.A.; Jouhet, J.; Meneghesso, A.; Valle, G.; Giuliano, G.; Maréchal, E. Light remodels lipid biosynthesis in Nannochloropsis gaditana by modulating carbon partitioning between organelles. Plant Physiol. 2016, 171, 2468–2482. [Google Scholar] [CrossRef]
- McKew, B.A.; Lefebvre, S.C.; Achterberg, E.P.; Metodieva, G.; Raines, C.A.; Metodiev, M.V.; Geider, R.J. Plasticity in the proteome of Emiliania huxleyi CCMP 1516 to extremes of light is highly targeted. New Phytol. 2013, 200, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Jia, J.; Li, J.; Sommerfeld, M.; Xu, J.; Hu, Q. Metabolic remodeling of membrane glycerolipids in the microalga Nannochloropsis oceanica under nitrogen deprivation. Front. Mar. Sci. 2017, 4, 242. [Google Scholar] [CrossRef]
- Janssen, J.H.; Lamers, P.P.; de Vos, R.C.; Wijffels, R.H.; Barbosa, M.J. Translocation and de novo synthesis of eicosapentaenoic acid (EPA) during nitrogen starvation in Nannochloropsis gaditana. Algal Res. 2019, 37, 138–144. [Google Scholar] [CrossRef]
- Yamashita, A.; Sugiura, T.; Waku, K. Acyltransferases and transacylases involved in fatty acid remodeling of phospholipids and metabolism of bioactive lipids in mammalian cells. J. Biochem. 1997, 122, 1–16. [Google Scholar] [CrossRef]
- Barbosa, M.; Valentão, P.; Andrade, P.B. Biologically active oxylipins from enzymatic and nonenzymatic routes in macroalgae. Mar. Drugs 2016, 14, 23. [Google Scholar] [CrossRef]
- De los Reyes, C.; Ávila-Román, J.; Ortega, M.J.; de la Jara, A.; García-Mauriño, S.; Motilva, V.; Zubía, E. Oxylipins from the microalgae Chlamydomonas debaryana and Nannochloropsis gaditana and their activity as TNF-α inhibitors. Phytochemistry 2014, 102, 152–161. [Google Scholar] [CrossRef]
- Longworth, J.; Wu, D.; Huete-Ortega, M.; Wright, P.C.; Vaidyanathan, S. Proteome response of Phaeodactylum tricornutum, during lipid accumulation induced by nitrogen depletion. Algal Res. 2016, 18, 213–224. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef]
- Adav, S.S.; Chao, L.T.; Sze, S.K. Protein abundance in multiplexed samples (PAMUS) for quantitation of Trichoderma reesei secretome. J. Proteom. 2013, 83, 180–196. [Google Scholar] [CrossRef]
- Ishihama, Y.; Oda, Y.; Tabata, T.; Sato, T.; Nagasu, T.; Rappsilber, J.; Mann, M. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteom. 2005, 4, 1265–1272. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Sayols, S.; Scherzinger, D.; Klein, H. dupRadar: A Bioconductor package for the assessment of PCR artifacts in RNA-Seq data. BMC Bioinform. 2016, 17, 428. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X. A survey of best practices for RNA-seq data analysis. Genome Biology 2016, 17, 13. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Alonso, R.; Salavert, F.; Garcia-Garcia, F.; Carbonell-Caballero, J.; Bleda, M.; Garcia-Alonso, L.; Sanchis-Juan, A.; Perez-Gil, D.; Marin-Garcia, P.; Sanchez, R. Babelomics 5.0: Functional interpretation for new generations of genomic data. Nucleic Acids Res. 2015, 43, W117–W121. [Google Scholar] [CrossRef]
- Luo, W.; Friedman, M.S.; Shedden, K.; Hankenson, K.D.; Woolf, P.J. GAGE: Generally applicable gene set enrichment for pathway analysis. BMC Bioinform. 2009, 10, 161. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. R Package Version 2. 2010. Available online: https://bioc.ism.ac.jp/packages/2.14/bioc/html/topGO.html (accessed on 20 September 2020).
- Pinheiro, J.C.; Bates, D.M. Mixed-Effects Models in S and S-PLUS; Springer: New York, NY, USA, 2000. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Identified Proteins | Accession Number | kDa | L2fc | p-Value |
|---|---|---|---|---|---|
| Upregulated | |||||
| 1 | Lipid droplet surface protein | W7TWF7_9STRA | 18 | 1.93 | 0.0001 |
| 2 | Amine oxidase | W7TFN3_9STRA | 75 | 1.38 | 0.0001 |
| 3 | Methylenetetrahydrofolate dehydrogenase | W7T6I6_9STRA | 39 | 1.3 | 0.0001 |
| 4 | Acid sphingomyelinase-like phosphodiesterase 3b | W7TQ09_9STRA | 76 | 1.3 | 0.001 |
| 5 | EF-Hand 1, calcium-binding site | W7TRW6_9STRA | 64 | 1.11 | 0.0001 |
| 6 | Lipase family protein | W7TUB0_9STRA | 54 | 1.06 | 0.0001 |
| 7 | Two component regulator propeller domain-containing protein | K8Z0G9_NANGC | 27 | 1.03 | 0.001 |
| 8 | Lipocalin protein | W7TQX7_9STRA | 29 | 1.02 | 0.00021 |
| 9 | Ammonium transporter | W7U477_9STRA | 58 | 1 | 0.0001 |
| 10 | Carbonic anhydrase, alpha-class | W7T0A1_9STRA | 37 | 0.9 | 0.028 |
| 11 | Cathepsin a | W7TYE0_9STRA | 60 | 0.87 | 0.0001 |
| 12 | Nadp-dependent glyceraldehyde-3-phosphate dehydrogenase | W7U8W3_9STRA | 66 | 0.86 | 0.0001 |
| 13 | Cluster of Sodium hydrogen exchanger 8 | W7TNK5_9STRA | 72 | 0.86 | 0.0001 |
| 14 | Light harvesting complex protein | K8YPR7_NANGC | 19 | 0.85 | 0.0001 |
| 15 | Subfamily member 9 | W7TPA4_9STRA | 41 | 0.82 | 0.028 |
| 16 | Plasma membrane ATPase | K8YQB4_NANGC | 107 | 0.77 | 0.0001 |
| 17 | Manganese lipoxygenase | W7TYD4_9STRA | 73 | 0.77 | 0.0001 |
| 18 | Quinoprotein amine dehydrogenase, beta chain | W7TI92_9STRA | 66 | 0.77 | 0.0001 |
| 19 | 4-hydroxyphenylpyruvate dioxygenase | W7TNB7_9STRA | 50 | 0.77 | 0.001 |
| 20 | Malate cytoplasmic isoform 2 | W7TPM0_9STRA | 37 | 0.76 | 0.0001 |
| 21 | Cluster of Violaxanthin de-epoxidase | K8YTT8_NANGC | 35 | 0.75 | 0.019 |
| 22 | Had-superfamily subfamily iia hydrolase | W7U270_9STRA | 43 | 0.74 | 0.0001 |
| 23 | Glutaryl-mitochondrial | W7TTQ4_9STRA | 48 | 0.74 | 0.0001 |
| 24 | Pyruvate dehydrogenase | W7TN62_9STRA | 55 | 0.74 | 0.0001 |
| 25 | Myotubularin-related protein 2 | W7TSB4_9STRA | 109 | 0.74 | 0.004 |
| 26 | Cdgsh iron sulfur domain-containing protein 1 | W7TPN8_9STRA | 23 | 0.72 | 0.001 |
| 27 | Arachidonate 5-lipoxygenase | K8Z8I5_NANGC | 60 | 0.71 | 0.0001 |
| 28 | Cluster of Purple acid phosphatase | W7TLQ2_9STRA | 56 | 0.71 | 0.0001 |
| 29 | Cluster of Expulsion defective family member (Exp-2) | K8YVZ3_NANGC | 62 | 0.71 | 0.049 |
| 30 | V-type proton ATPase subunit F | W7TU11_9STRA | 13 | 0.7 | 0.0001 |
| Downregulated | |||||
| 30 | Cytochrome p450 | W7UBA8_9STRA | 70 | −0.77 | 0.0001 |
| 29 | 30s ribosomal protein s15 | W7TEF2_9STRA | 34 | −0.77 | 0.0001 |
| 28 | RNA binding s1 domain protein | W7U882_9STRA | 45 | −0.77 | 0.0001 |
| 27 | Cluster of Solute carrier family 35 member b1 | W7TCR9_9STRA | 43 | −0.77 | 0.7 |
| 26 | Cytochrome P450 enzyme | I2CNY8_NANGC | 67 | −0.78 | 0.001 |
| 25 | Heat shock protein DNAJ, cysteine-rich domain protein | W7TJ91_9STRA | 13 | −0.78 | 0.001 |
| 24 | Geranylgeranyl reductase | W7THD6_9STRA | 57 | −0.79 | 0.0001 |
| 23 | Coproporphyrinogen iii oxidase chloroplast | W7TZ92_9STRA | 46 | −0.79 | 0.0001 |
| 22 | 50S ribosomal protein L18, chloroplastic | K9ZX62_9STRA | 12 | −0.8 | 0.0001 |
| 21 | 50S ribosomal protein L19 | K9ZV73_9STRA | 14 | −0.81 | 0.0001 |
| 20 | 30S ribosomal protein S9, chloroplastic | A0A023PLK7_9STRA | 15 | −0.82 | 0.0001 |
| 19 | 30S ribosomal protein S2, chloroplastic | K9ZWC8_9STRA | 29 | −0.83 | 0.0001 |
| 18 | Nitrite reductase | W7T0E9_9STRA | 46 | −0.85 | 0.0001 |
| 17 | 30S ribosomal protein S8, chloroplastic | K9ZV68_9STRA | 15 | −0.86 | 0.0001 |
| 16 | Cluster of H+-transporting ATPase | K8YQ29_NANGC | 152 | −0.87 | 0.0001 |
| 15 | 30S ribosomal protein S12, chloroplastic | K9ZVC5_9STRA | 14 | −0.88 | 0.0001 |
| 14 | 50S ribosomal protein L36, chloroplastic | K9ZXS5_9STRA | 4 | −0.88 | 0.001 |
| 13 | Magnesium chelatase ATPase subunit I | K9ZV21_9STRA | 47 | −0.9 | 0.0001 |
| 12 | 50S ribosomal protein L16, chloroplastic | K9ZWF3_9STRA | 16 | −0.9 | 0.0001 |
| 11 | Ribosomal protein s21 | W7TSY1_9STRA | 14 | −0.9 | 0.003 |
| 10 | Cluster of Mfs transporter | W7U968_9STRA | 66 | −0.93 | 0.14 |
| 9 | 30S ribosomal protein S17, chloroplastic | K9ZVE6_9STRA | 10 | −0.94 | 0.0001 |
| 8 | 30S ribosomal protein S20, chloroplastic | K9ZX69_9STRA | 11 | −0.94 | 0.0001 |
| 7 | Delta 5 fatty acid desaturase | K8YSX2_NANGC | 54 | −0.95 | 0.0001 |
| 6 | 30S ribosomal protein S18, chloroplastic | K9ZV97_9STRA | 8 | −0.97 | 0.0001 |
| 5 | Nitrate reductase | W7TAR6_9STRA | 70 | −1.08 | 0.0001 |
| 4 | Ferredoxin nitrite reductase | K8YST4_NANGC | 40 | −1.13 | 0.0001 |
| 3 | Light-independent protochlorophyllide reductase subunit N | K9ZV79_9STRA | 50 | −1.15 | 0.0001 |
| 2 | Light-independent protochlorophyllide reductase iron-sulfur ATP-binding protein | K9ZV32_9STRA | 32 | −1.34 | 0.0001 |
| 1 | NAD(P)H nitrate reductase | K8YSU6_NANGC | 63 | −1.48 | 0.0001 |
| Rank | Identified Proteins | Accession Number | kDa | L2fc | p-Value |
|---|---|---|---|---|---|
| Upregulated | |||||
| 1 | Sse2p | W7TMT9_9STRA | 32 | 0.96 | 0.04 |
| 2 | Acid sphingomyelinase-like phosphodiesterase 3b | W7TQ09_9STRA | 76 | 0.68 | 0.011 |
| 3 | Cluster of Calcium binding protein 39 | W7T646_9STRA | 51 | 0.61 | 0.59 |
| 4 | Snf7 family protein | W7U1R3_9STRA | 22 | 0.53 | 0.026 |
| 5 | Ddi1p | W7U1J9_9STRA | 41 | 0.52 | 0.97 |
| 6 | Nad-dependent deacetylase | W7TT51_9STRA | 38 | 0.48 | 0.004 |
| 7 | Elongation of fatty acids protein | W7TSM8_9STRA | 36 | 0.48 | 0.06 |
| 8 | Lysyl-tRNA synthetase | W7TMK7_9STRA | 20 | 0.45 | 0.13 |
| 9 | Aminoglycoside phosphotransferase | W7TK75_9STRA | 37 | 0.43 | 0.2 |
| 10 | Pyruvate decarboxylase | K8YS66_NANGC | 62 | 0.41 | 0.0001 |
| 11 | Splicing arginine serine-rich 19 | W7T8W4_9STRA | 34 | 0.41 | 0.0001 |
| 12 | Ribosomal protein | K8Z5W4_NANGC | 33 | 0.39 | 0.067 |
| 13 | Cluster of Trypsin family | K8Z6K0_NANGC | 65 | 0.38 | 0.36 |
| 14 | Cluster of Methylthioribose kinase | W7TVE0_9STRA | 94 | 0.37 | 0.98 |
| 15 | Ferredoxin | K8YW46_NANGC | 12 | 0.36 | 0.055 |
| 16 | Otu-like cysteine type protease | W7TUL0_9STRA | 102 | 0.36 | 0.15 |
| 17 | Protein-tyrosine low molecular weight | K8YTE7_NANGC | 16 | 0.35 | 0.00023 |
| 18 | Threonine aldolase | W7TQZ9_9STRA | 47 | 0.35 | 0.012 |
| 19 | Protein phosphatase | W7TA28_9STRA | 48 | 0.35 | 0.2 |
| 20 | Pre-mRNA-processing factor 17 | K8Z4U6_NANGC | 86 | 0.34 | 0.0001 |
| 21 | Beta-ketoacyl-thiolase | W7SYP3_9STRA | 8 | 0.34 | 0.022 |
| 22 | Ethylmalonic encephalopathy 1 | K8Z7T8_NANGC | 47 | 0.33 | 0.0001 |
| 23 | Soluble pyridine nucleotide transhydrogenase | W7T7X5_9STRA | 17 | 0.32 | 0.14 |
| 24 | Ring-finger-containing e3 ubiquitin | W7UAK3_9STRA | 76 | 0.32 | 0.25 |
| 25 | Glycerol kinase | W7U0M7_9STRA | 24 | 0.3 | 0.072 |
| 26 | Ig family protein | W7T9Y3_9STRA | 60 | 0.3 | 0.082 |
| 27 | Cluster of Mfs transporter | W7U968_9STRA | 66 | 0.3 | 0.89 |
| 28 | Mitochondrial tricarboxylate carrier family | W7TKI7_9STRA | 36 | 0.29 | 0.009 |
| 29 | Cdgsh iron sulfur domain-containing protein 1 | W7TPN8_9STRA | 23 | 0.29 | 0.025 |
| 30 | NAD(P)-binding domain protein | W7TM45_9STRA | 40 | 0.29 | 0.032 |
| Downregulated | |||||
| 30 | Vacuolar protein-sorting-associated protein 36 | W7TG31_9STRA | 49 | −0.44 | 0.34 |
| 29 | Exocyst complex | W7U8I8_9STRA | 115 | −0.45 | 0.009 |
| 28 | Methyltransferase type 11 | W7U3Q9_9STRA | 34 | −0.45 | 0.027 |
| 27 | RNA binding protein | W7TAT7_9STRA | 20 | −0.47 | 0.29 |
| 26 | Light harvesting complex protein | K8YPR7_NANGC | 19 | −0.5 | 0.007 |
| 25 | Diaminopimelate decarboxylase | W7TNX0_9STRA | 56 | −0.5 | 0.04 |
| 24 | DNA polymerase subunit Cdc27 | W7TMW3_9STRA | 62 | −0.51 | 0.013 |
| 23 | Tubulin-tyrosine ligase-like protein | W7TWY1_9STRA | 79 | −0.51 | 0.024 |
| 22 | Translocase of inner mitochondrial membrane 50-like protein | K8YTV0_NANGC | 43 | −0.51 | 0.13 |
| 21 | Cluster of Protease do-like 9 | W7TU24_9STRA | 69 | −0.52 | 0.61 |
| 20 | TatA-like sec-independent protein translocator subunit | W7T3A7_9STRA | 22 | −0.54 | 0.001 |
| 19 | Photosystem II reaction center protein H | K9ZXQ7_9STRA | 7 | −0.54 | 0.084 |
| 18 | Cyclic nucleotide-binding protein | W7TMP7_9STRA | 25 | −0.56 | 0.002 |
| 17 | Ubiquilin | I2CQX3_NANGC | 47 | −0.6 | 0.07 |
| 16 | Ribokinase | W7TXK5_9STRA | 34 | −0.62 | 0.21 |
| 15 | Ankyrin | W7TWU3_9STRA | 48 | −0.63 | 0.068 |
| 14 | Soluble nsf attachment protein receptor | W7TW41_9STRA | 32 | −0.66 | 0.27 |
| 13 | Elongation of fatty acids protein | W7U1Y8_9STRA | 37 | −0.67 | 0.098 |
| 12 | Anamorsin homolog | W7TKP2_9STRA | 30 | −0.67 | 0.19 |
| 11 | Adenylate kinase | K8ZCS9_NANGC | 19 | −0.68 | 0.1 |
| 10 | Mitochondrial carrier domain protein | W7TRC0_9STRA | 50 | −0.68 | 0.13 |
| 9 | Set domain protein | W7TKH2_9STRA | 119 | −0.73 | 0.75 |
| 8 | ATP-dependent RNA helicase DDX23/PRP28 | K8YWH1_NANGC | 91 | −0.77 | 0.13 |
| 7 | Pentatricopeptide repeat-containing protein | W7TSL2_9STRA | 138 | −0.81 | 0.24 |
| 6 | Fgd6 protein | K8Z5M8_NANGC | 33 | −0.84 | 0.26 |
| 5 | Polypyrimidine tract binding protein | I2CQY0_NANGC | 35 | −0.96 | 0.01 |
| 4 | Major facilitator superfamily | W7UAL7_9STRA | 66 | −0.99 | 0.21 |
| 3 | U3 small nucleolar RNA-associated | W7UBP4_9STRA | 207 | −1.1 | 0.049 |
| 2 | Phytanoyl-dioxygenase | W7T3Z1_9STRA | 24 | −1.2 | 0.047 |
| 1 | DNA damage-binding protein 1a | I2CQY4_NANGC | 41 | −1.45 | 0.14 |
| Day 3 | Day 5 | ||||
|---|---|---|---|---|---|
| Treatment | No. Cultivations | Protein | Transcript | Protein | Transcript |
| Control (C) | 4 | 4 | 4 | − | 4 |
| Nitrogen (N-) | 4 | 4 | 4 | − | 4 |
| Phosphorus (P-) | 4 | 2 | 4 | − | 4 |
| Total | 12 | 10 | 12 | − | 12 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hulatt, C.J.; Smolina, I.; Dowle, A.; Kopp, M.; Vasanth, G.K.; Hoarau, G.G.; Wijffels, R.H.; Kiron, V. Proteomic and Transcriptomic Patterns during Lipid Remodeling in Nannochloropsis gaditana. Int. J. Mol. Sci. 2020, 21, 6946. https://doi.org/10.3390/ijms21186946
Hulatt CJ, Smolina I, Dowle A, Kopp M, Vasanth GK, Hoarau GG, Wijffels RH, Kiron V. Proteomic and Transcriptomic Patterns during Lipid Remodeling in Nannochloropsis gaditana. International Journal of Molecular Sciences. 2020; 21(18):6946. https://doi.org/10.3390/ijms21186946
Chicago/Turabian StyleHulatt, Chris J., Irina Smolina, Adam Dowle, Martina Kopp, Ghana K. Vasanth, Galice G. Hoarau, René H. Wijffels, and Viswanath Kiron. 2020. "Proteomic and Transcriptomic Patterns during Lipid Remodeling in Nannochloropsis gaditana" International Journal of Molecular Sciences 21, no. 18: 6946. https://doi.org/10.3390/ijms21186946
APA StyleHulatt, C. J., Smolina, I., Dowle, A., Kopp, M., Vasanth, G. K., Hoarau, G. G., Wijffels, R. H., & Kiron, V. (2020). Proteomic and Transcriptomic Patterns during Lipid Remodeling in Nannochloropsis gaditana. International Journal of Molecular Sciences, 21(18), 6946. https://doi.org/10.3390/ijms21186946