Contribution of Autophagy-Notch1-Mediated NLRP3 Inflammasome Activation to Chronic Inflammation and Fibrosis in Keloid Fibroblasts

 , , and
, , and 
Abstract
:1. Introduction
2. Results
2.1. Clinical Characteristics of Patients with Keloid
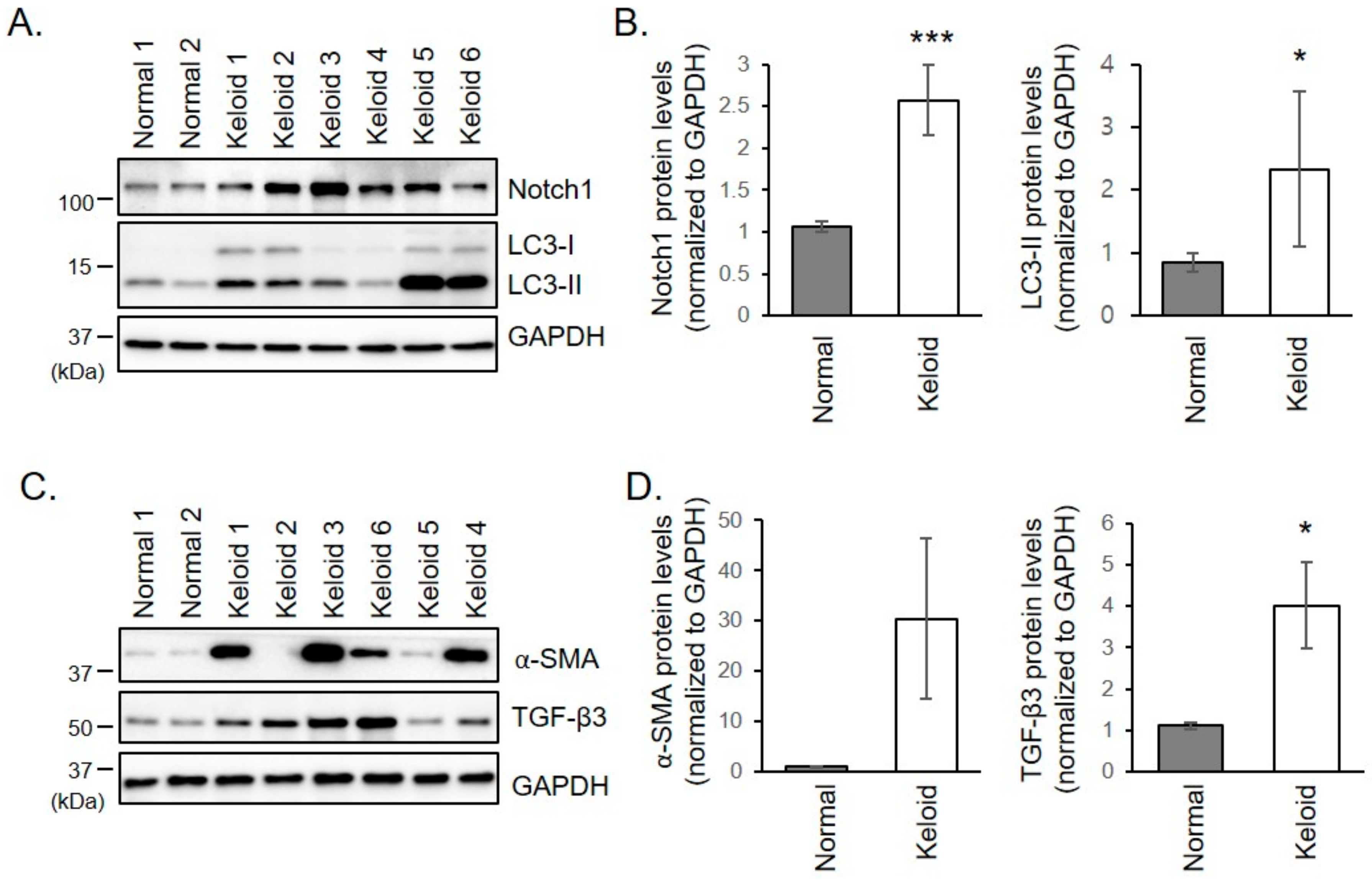
2.2. Notch1, α-SMA, and TGF-β3 Protein Levels are Increased in Keloid Fibroblasts
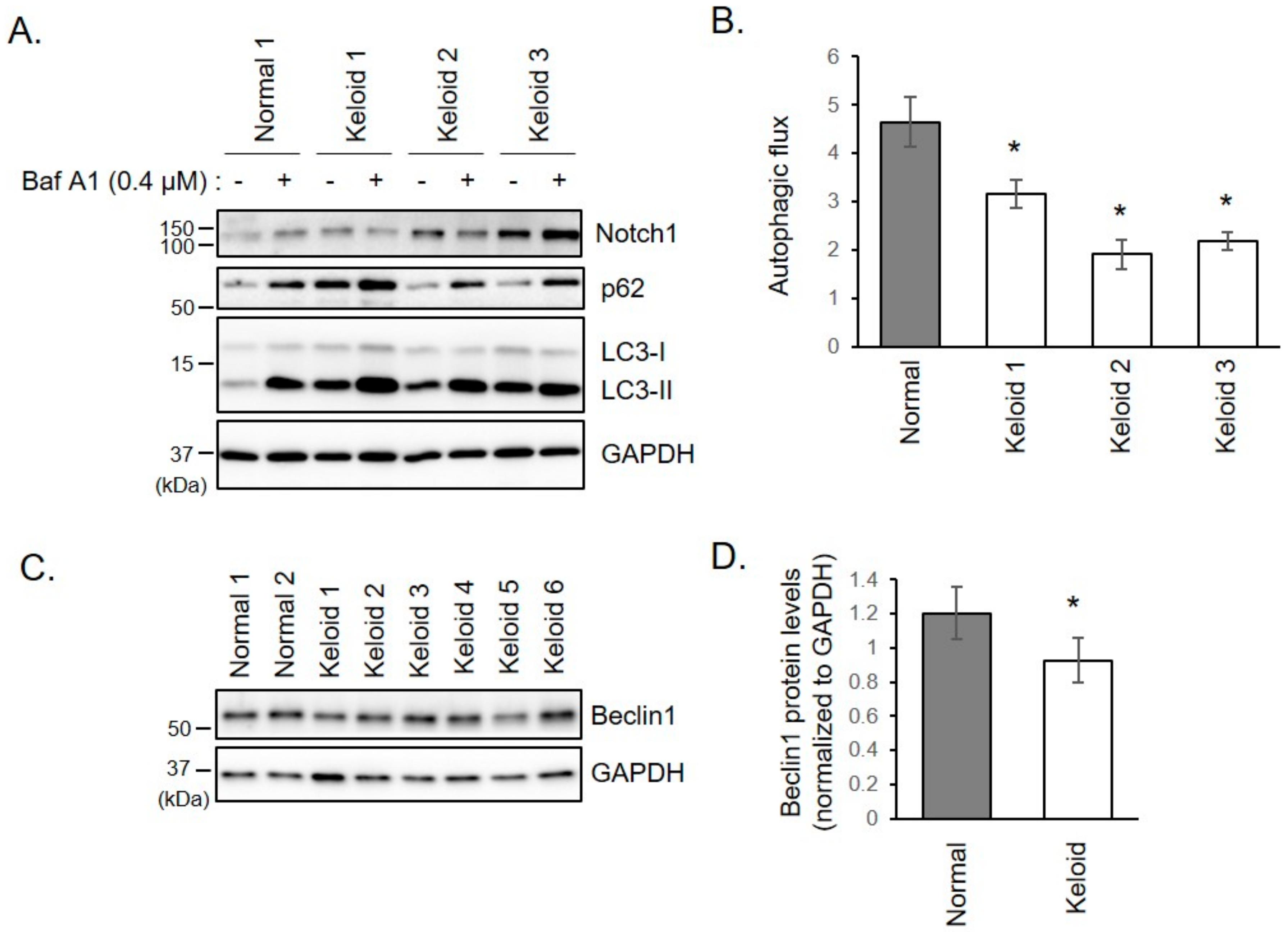
2.3. Autophagy is Disturbed in Keloid Fibroblasts
2.4. NLRP3 Inflammasome Formation is Activated in Keloid Fibroblasts
2.5. Notch Inhibition with siRNA Down-Regulates the Activation of NLRP3 Inflammasome Formation and the Differentiation into Myofibroblasts
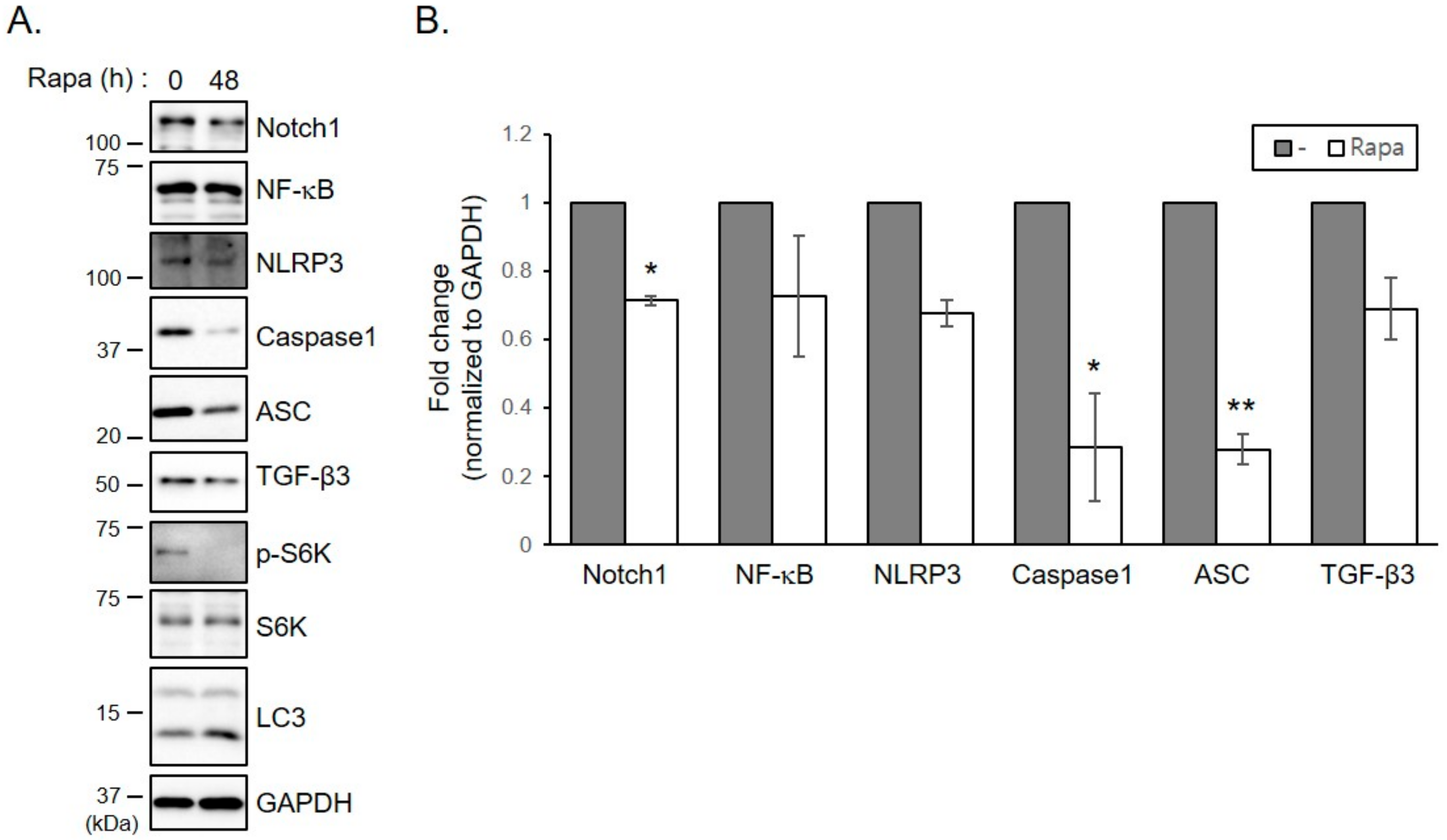
2.6. Treatment of Keloid Fibroblasts with Rapamycin Reduces Increased Levels of Inflammasomes and TGF- β3 Proteins
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Cell Culture
4.3. Quantitative Real-Time PCR (qRT-PCR)
4.4. Western Blot Analysis
4.5. siRNA Transfection
4.6. Drug Treatment
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| α-SMA | Alpha-smooth muscle actin |
| DAMP | Damage-associated molecular pattern |
| ECM | Extracellular matrix |
| HMGB1 | High mobility group box 1 |
| IL | Interleukin |
| mTOR | Mammalian target of rapamycin |
| NF-κB | Nuclear factor kappa B |
| NICD | Notch intracellular domain |
| NLRP3 | NACHT, LRR, and PYD domains-containing protein 3 |
| PAMP | Pathogen-associated molecular pattern |
| PCNA | Proliferating cell nuclear antigen |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| TGF-β | Transforming growth factor beta |
| TLR | Toll-like receptor |
References
- Ogawa, R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int. J. Mol. Sci. 2017, 18, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, B.; Maderal, A.; Raphael, B. Keloids and Hypertrophic Scars: Pathophysiology, Classification, and Treatment. Dermatol. Surg. 2017, 43, S3–S18. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Lee, J.-H.; Jeong, K.H.; Kim, G.M.; Kang, H. Notch intracellular domain expression in various skin fibroproliferative diseases. Ann. Dermatol. 2014, 26, 332–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, F.; Hernandez, R.M. Notch signaling pathway in keloid disease: Enhanced fibroblast activity in a Jagged-1 peptide-dependent manner in lesional vs. extralesional fibroblasts. Wound Repair Regen. 2012, 20, 688–706. [Google Scholar] [CrossRef] [PubMed]
- Kavian, N.; Servettaz, A.; Weill, B.; Batteux, F. New insights into the mechanism of notch signalling in fibrosis. Open Rheumatol. J. 2012, 6, 96–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstädt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef] [Green Version]
- Dees, C.; Tomcik, M.; Zerr, P.; Akhmetshina, A.; Horn, A.; Palumbo, K.; Beyer, C.; Zwerina, J.; Distler, O.; Schett, G.; et al. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Ann. Rheum. Dis. 2011, 70, 1304–1310. [Google Scholar] [CrossRef] [Green Version]
- Aoyagi-Ikeda, K.; Maeno, T.; Matsui, H.; Ueno, M.; Hara, K.; Aoki, Y.; Aoki, F.; Shimizu, T.; Doi, H.; Kawai-Kowase, K.; et al. Notch induces myofibroblast differentiation of alveolar epithelial cells via transforming growth factor-{beta}-Smad3 pathway. Am. J. Respir. Cell Mol. Biol. 2011, 45, 136–144. [Google Scholar]
- Jun, J.-I.; Lau, L.F. Cellular senescence controls fibrosis in wound healing. Aging 2010, 2, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Park, H.; Kang, J.-H.; Lee, S. Autophagy in neurodegenerative diseases: A hunter for aggregates. Int. J. Mol. Sci. 2020, 21, 3369. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Fleming, A.; Ricketts, T.; Pavel, M.; Virgin, H.; Menzies, F.M.; Rubinsztein, D.C. Autophagy regulates notch degradation and modulates stem cell development and neurogenesis. Nat. Commun. 2016, 7, 10533. [Google Scholar] [CrossRef] [Green Version]
- Rybstein, M.D.; Bravo-San Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. The autophagic network and cancer. Nat. Cell Biol. 2018, 20, 243–251. [Google Scholar] [CrossRef]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, H.; Kanbe, A.; Sakai, H.; Seishima, M. Activation of NLRP3 signaling accelerates skin wound healing. Exp. Dermatol. 2017, 27, 80–86. [Google Scholar] [CrossRef]
- Lv, Z.; Wang, Y.; Liu, Y.J.; Mao, Y.F.; Dong, W.-W.; Ding, Z.-N.; Meng, G.-X.; Jiang, L.; Zhu, X.-Y. NLRP3 inflammasome activation contributes to mechanical stretch–induced endothelial-mesenchymal transition and pulmonary fibrosis. Crit. Care Med. 2018, 46, e49–e58. [Google Scholar] [CrossRef] [PubMed]
- Torp, M.K.; Yang, K.; Ranheim, T.; Huso Lauritzen, K.; Alfsnes, K.; Vinge, L.E.; Aukrust, P.; Stenslokken, K.O.; Yndestad, A.; Sandanger, O. Mammalian target of rapamycin (mTOR) and the proteasome attenuates IL-1beta expression in primary mouse cardiac fibroblasts. Front. Immunol. 2019, 10, 1285. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.-C.; Liu, Y.; Cen, Y.-Y.; Xiong, Y.-L.; Li, J.-M.; Ding, Y.-Y.; Tong, Y.-F.; Liu, T.; Chen, X.-H.; Zhang, H.-G. Dual role of triptolide in interrupting the NLRP3 inflammasome pathway to attenuate cardiac fibrosis. Int. J. Mol. Sci. 2019, 20, 360. [Google Scholar] [CrossRef] [Green Version]
- Bernard, M.; Dieudé, M.; Yang, B.; Hamelin, K.; Underwood, K.; Hébert, M.-J. Autophagy fosters myofibroblast differentiation through MTORC2 activation and downstream upregulation of CTGF. Autophagy 2014, 10, 2193–2207. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Li, W.; Wen, J.; Yang, Z. Autophagy is involved in mouse kidney development and podocyte differentiation regulated by Notch signalling. J. Cell. Mol. Med. 2017, 21, 1315–1328. [Google Scholar] [CrossRef]
- Nam, S.A.; Kim, W.-Y.; Kim, J.W.; Park, S.H.; Kim, H.L.; Lee, M.-S.; Komatsu, M.; Ha, H.; Lim, J.H.; Park, C.W.; et al. Autophagy attenuates tubulointerstital fibrosis through regulating transforming growth factor-β and NLRP3 inflammasome signaling pathway. Cell Death Dis. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Seveau, S.; Turner, J.; Gavrilin, M.A.; Torrelles, J.B.; Hall-Stoodley, L.; Yount, J.S.; Amer, A.O. Checks and balances between autophagy and inflammasomes during infection. J. Mol. Biol. 2018, 430, 174–192. [Google Scholar] [CrossRef] [Green Version]
- El Ayadi, A.; Jay, J.W.; Prasai, A. Current approaches targeting the wound healing phases to attenuate fibrosis and scarring. Int. J. Mol. Sci. 2020, 21, 1105. [Google Scholar] [CrossRef] [Green Version]
- Ong, C.T.; Khoo, Y.T.; Mukhopadhyay, A.; Do, D.V.; Lim, I.J.; Aalami, O.; Phan, T.T. mTOR as a potential therapeutic target for treatment of keloids and excessive scars. Exp. Dermatol. 2007, 16, 394–404. [Google Scholar] [CrossRef]
- Lei, R.; Li, J.; Liu, F.; Li, W.; Zhang, S.; Wang, Y.; Chu, X.; Xu, J. HIF-1alpha promotes the keloid development through the activation of TGF-beta/Smad and TLR4/MyD88/NF-kappaB pathways. Cell Cycle 2019, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Li, J.; Liu, H.; Jian, X.; Zou, Q.; Zhao, Q.; Le, Q.; Chen, H.; Gao, X.; He, C. Adiponectin is involved in connective tissue growth factor-induced proliferation, migration and overproduction of the extracellular matrix in keloid fibroblasts. Int. J. Mol. Sci. 2017, 18, 1044. [Google Scholar] [CrossRef] [Green Version]
- Frings, V.G.; Sennefelder, H.; Presser, D.; Goebeler, M.; Schmidt, M. Altered NOX expression does not seem to account for epidermal NLRP 3 inflammasome activation in hidradenitis suppurativa. Br. J. Dermatol. 2019, 181, 391–392. [Google Scholar] [CrossRef]
- Marasca, C.; Scala, E.; Di Caprio, R.; Raimondo, A.; Cacciapuoti, S.; Balato, N.; Fabbrocini, G. Notch dysregulation and hidradenitis suppurativa, psoriasis, atopic dermatitis and lichen planus: Let’s talk about Numb. Br. J. Dermatol. 2019, 180, 950–951. [Google Scholar] [CrossRef] [PubMed]
- Do, D.; Ong, C.; Khoo, Y.; Carbone, A.; Lim, C.; Wang, S.; Mukhopadhyay, A.; Cao, X.; Cho, D.; Wei, X.; et al. Interleukin-18 system plays an important role in keloid pathogenesis via epithelial-mesenchymal interactions. Br. J. Dermatol. 2012, 166, 1275–1288. [Google Scholar] [CrossRef]
- Hou, J.; Ma, T.; Cao, H.; Chen, Y.; Wang, C.; Chen, X.; Xiang, Z.; Han, X. TNF-α-induced NF-κB activation promotes myofibroblast differentiation of LR-MSCs and exacerbates bleomycin-induced pulmonary fibrosis. J. Cell. Physiol. 2017, 233, 2409–2419. [Google Scholar] [CrossRef]
- Espinosa, L.; Cathelin, S.; D’Altri, T.; Trimarchi, T.; Statnikov, A.; Guiu, J.; Rodilla, V.; Inglés-Esteve, J.; Nomdedeu, J.; Bellosillo, B.; et al. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell Leukemia. Cancer Cell 2010, 18, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiu, Y.; Dong, Q.; Fu, L.; Bossler, A.; Tang, X.; Boyce, B.; Borcherding, N.; Leidinger, M.; Sardina, J.L.; Xue, H.-H.; et al. Coactivation of NF-κB and Notch signaling is sufficient to induce B-cell transformation and enables B-myeloid conversion. Blood 2020, 135, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Zhou, L.; Ochoa, S.; Hasan, A.; Badiavas, E.; Falanga, V. TGF-beta3 stimulates and regulates collagen synthesis through TGF-beta1-dependent and independent mechanisms. J. Investig. Dermatol. 1997, 108, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komai, T.; Okamura, T.; Inoue, M.; Yamamoto, K.; Fujio, K. Reevaluation of pluripotent Cytokine TGF-β3 in Immunity. Int. J. Mol. Sci. 2018, 19, 2261. [Google Scholar] [CrossRef] [Green Version]
- Lichtman, M.K.; Otero-Viñas, M.; Falanga, V. Transforming growth factor beta (TGF-β) isoforms in wound healing and fibrosis. Wound Repair Regen. 2016, 24, 215–222. [Google Scholar] [CrossRef]
- Jiang, L.; Ke, M.; Yue, S.; Xiao, W.; Yan, Y.; Deng, X.; Ying, Q.-L.; Li, J.; Ke, B. Blockade of notch signaling promotes acetaminophen-induced liver injury. Immunol. Res. 2017, 65, 739–749. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Keloid1 | Keloid2 | Keloid3 | Keloid4 | Keloid5 | Keloid6 | |
|---|---|---|---|---|---|---|
| Gender/age | F/26 | F/25 | F/65 | F/20 | F/18 | F/23 |
| Disease duration | 1 yr | 2 yrs | 3 yrs | 3 yrs | 1 yr | 2 yrs |
| Symptoms | (−) | Pain, pruritus Size increasing | Pain, pruritus Size increasing | (−) | Pain, pruritus Size increasing | (−) |
| Clinical findings |  |  |  | Not available |  |  |
| Previous treatment | No | ILI 1 month before excision | ILI 4 months before excision | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.; Kim, S.K.; Park, H.; Lee, Y.J.; Park, S.H.; Lee, K.J.; Lee, D.G.; Kang, H.; Kim, J.E. Contribution of Autophagy-Notch1-Mediated NLRP3 Inflammasome Activation to Chronic Inflammation and Fibrosis in Keloid Fibroblasts. Int. J. Mol. Sci. 2020, 21, 8050. https://doi.org/10.3390/ijms21218050
Lee S, Kim SK, Park H, Lee YJ, Park SH, Lee KJ, Lee DG, Kang H, Kim JE. Contribution of Autophagy-Notch1-Mediated NLRP3 Inflammasome Activation to Chronic Inflammation and Fibrosis in Keloid Fibroblasts. International Journal of Molecular Sciences. 2020; 21(21):8050. https://doi.org/10.3390/ijms21218050
Chicago/Turabian StyleLee, Seongju, Sun Kyeon Kim, Hyungsun Park, Yu Jin Lee, Song Hee Park, Kyung Jae Lee, Dong Geon Lee, Hoon Kang, and Jung Eun Kim. 2020. "Contribution of Autophagy-Notch1-Mediated NLRP3 Inflammasome Activation to Chronic Inflammation and Fibrosis in Keloid Fibroblasts" International Journal of Molecular Sciences 21, no. 21: 8050. https://doi.org/10.3390/ijms21218050