Dual-Inhibitors of N-Myc and AURKA as Potential Therapy for Neuroendocrine Prostate Cancer

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Rational Design of Novel Potent N-Myc Inhibitors
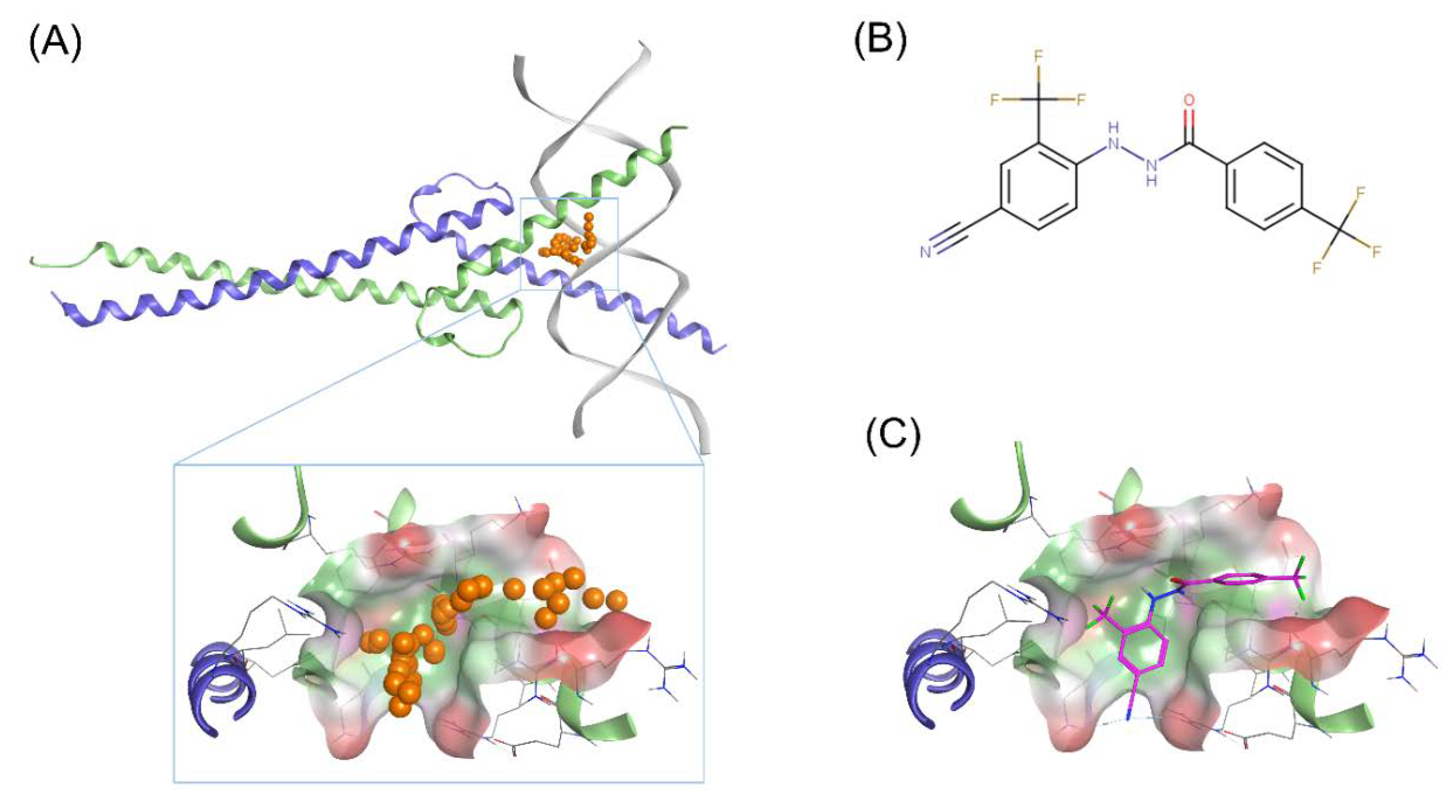
2.1.1. Homology Modelling of N-Myc-Max Heterodimer
2.1.2. Identification of Scaffolds Targeting the N-Myc-Max DBD Site
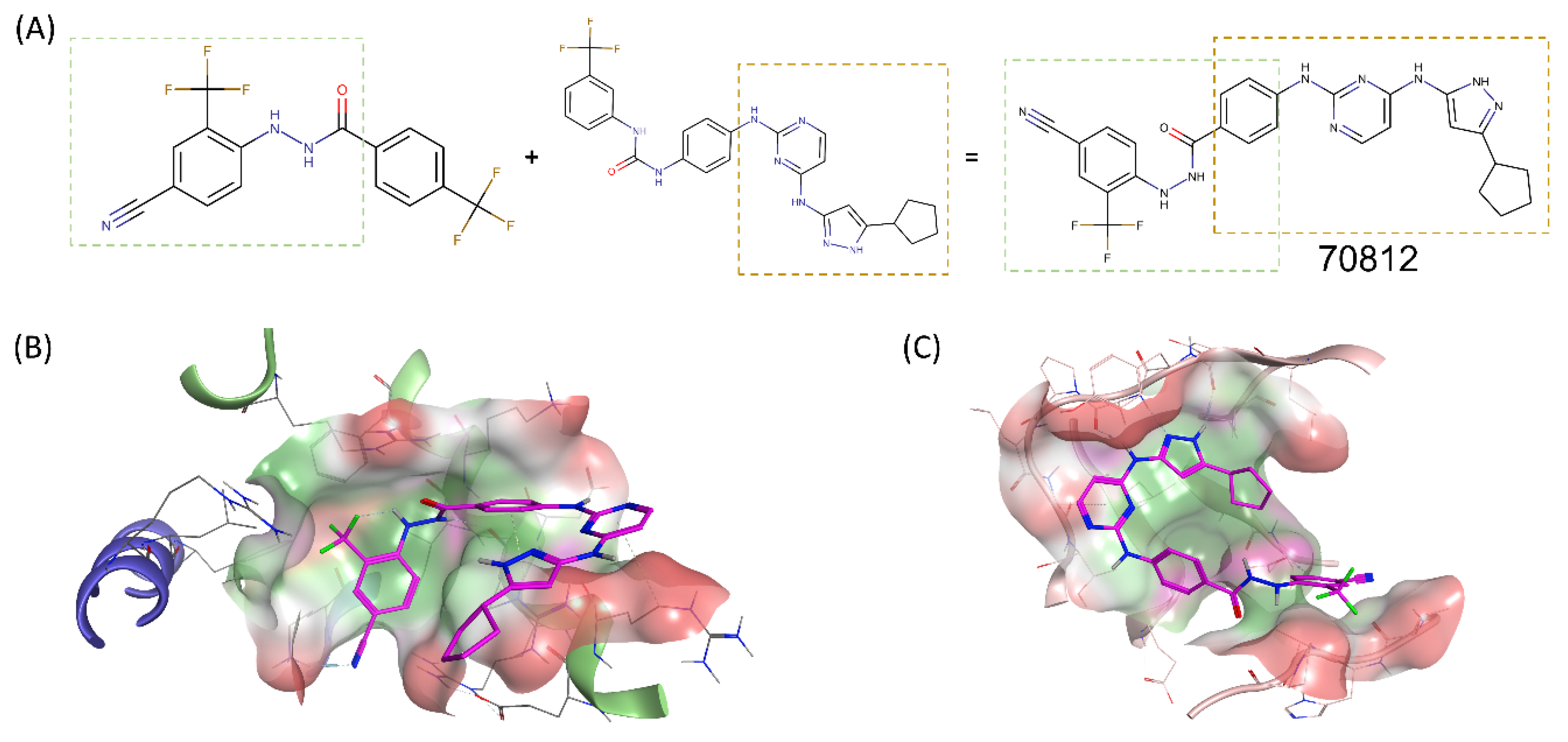
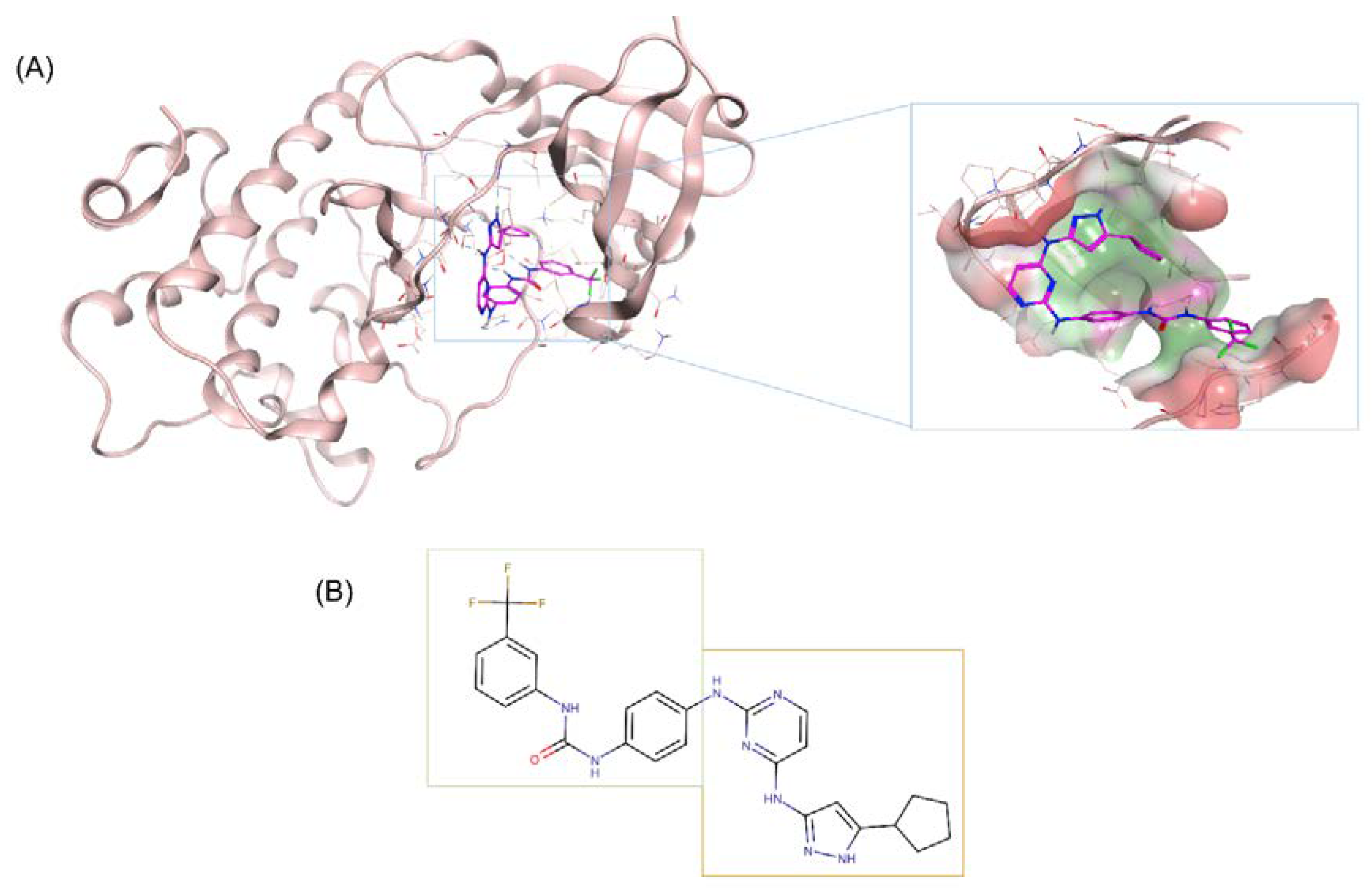
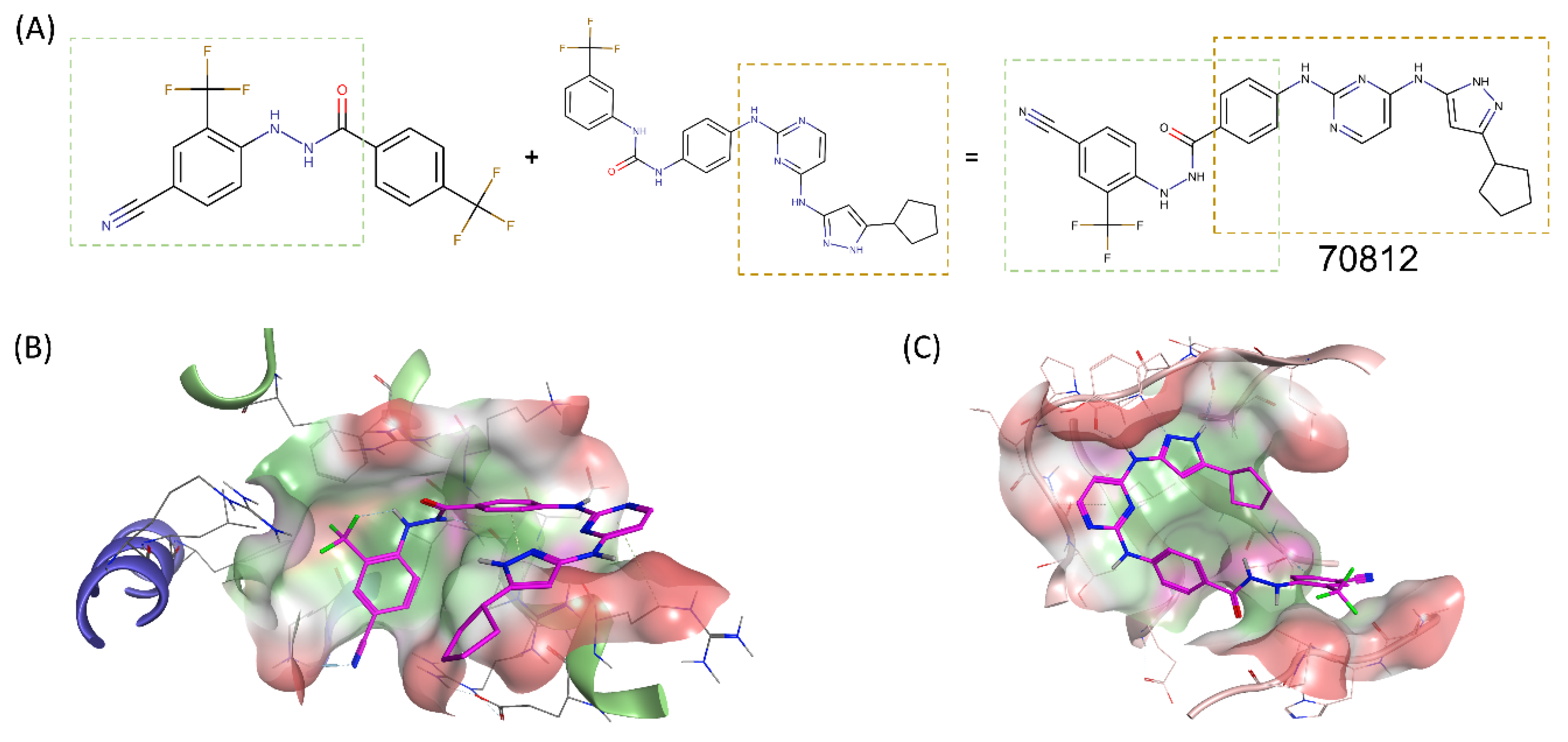
2.2. Structure-Based Drug Design of N-Myc and AURKA Dual-Inhibitors
2.3. Binding Prediction of 70812 to AURKA and N-Myc through Docking
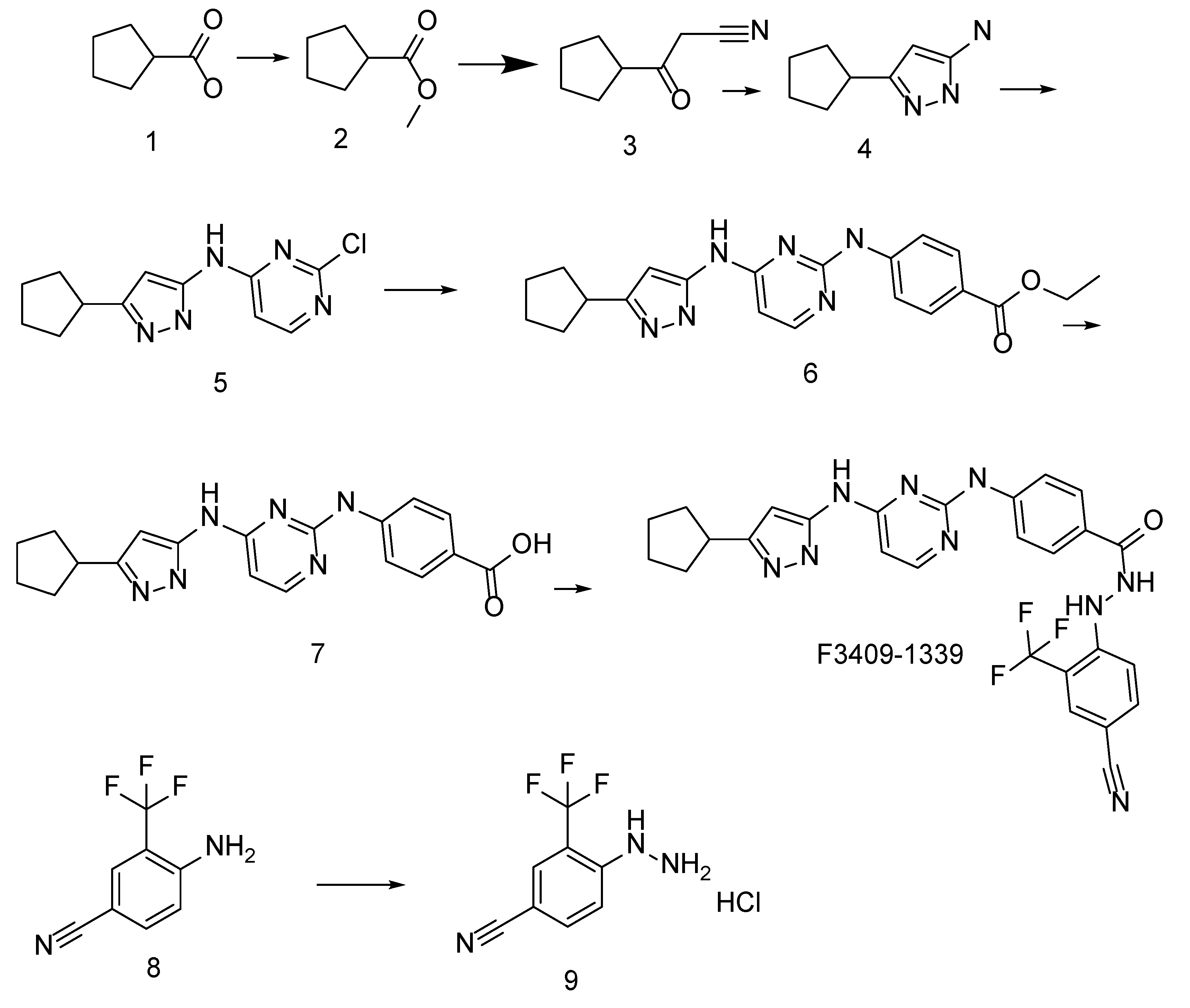
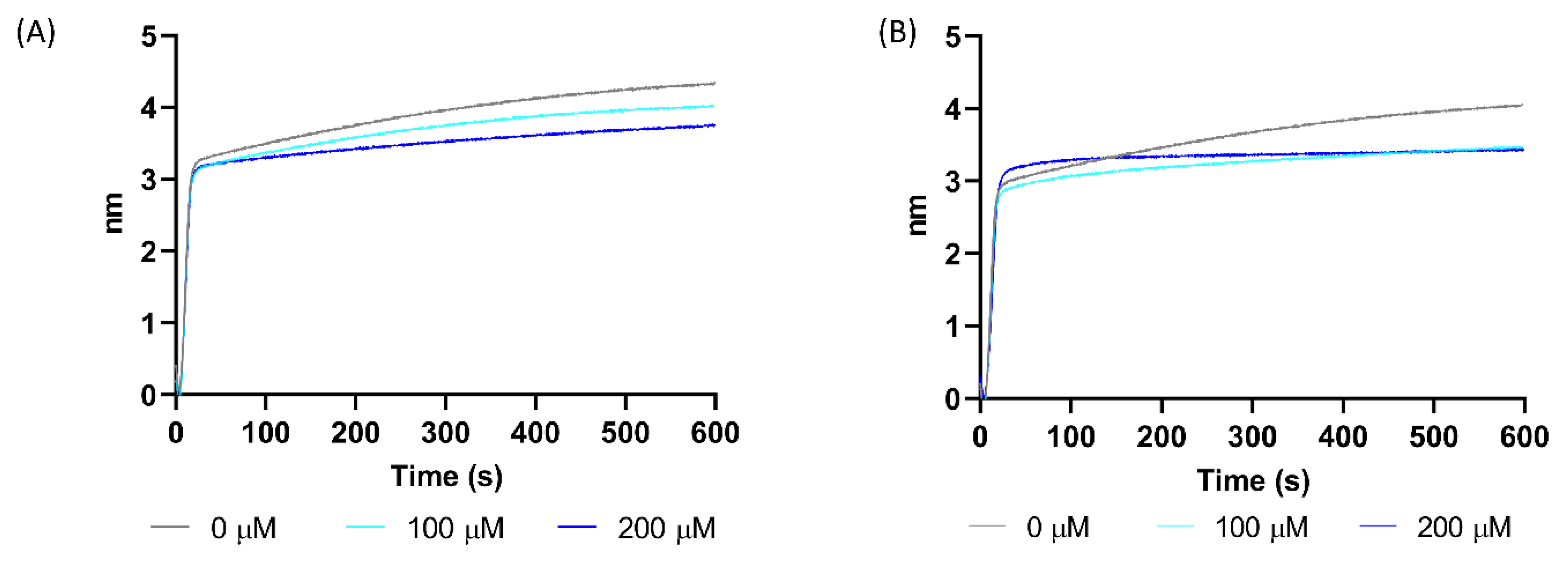
2.4. Experimental Validation of 70812 As Dual-Inhibitor
N′-[4-cyano-2-(trifluoromethyl)phenyl]-4-(trifluoromethyl)benzohydrazide as Active Scaffold against N-Myc
2.5. Biological Characterization of 70812 as a Dual-Inhibitor
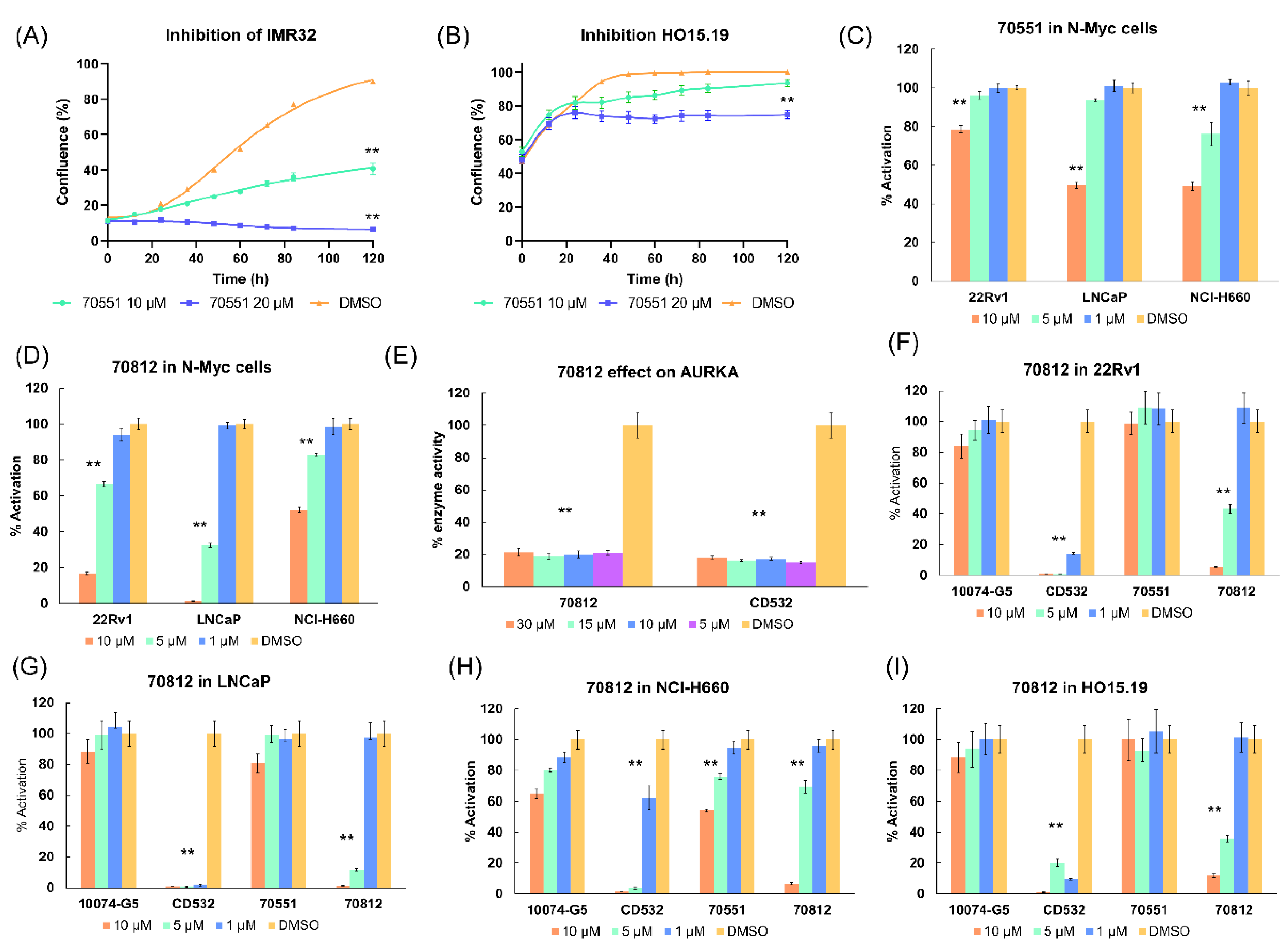
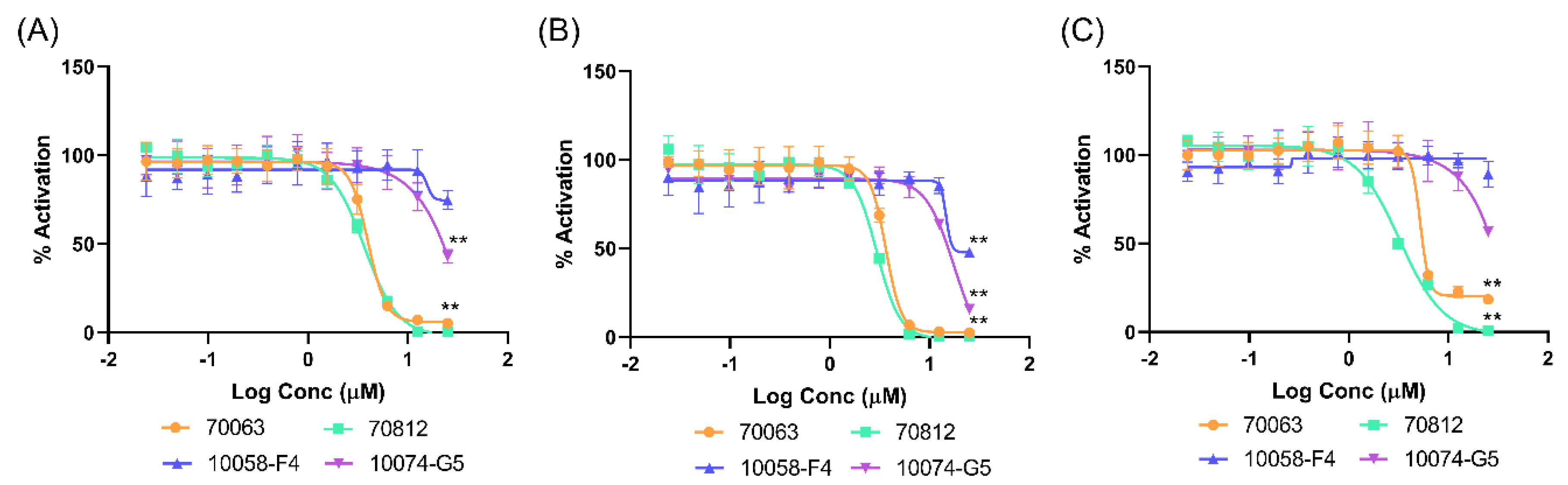
2.5.1. 70812 Is a Potent Inhibitor of Both N-Myc and AURKA
2.5.2. 70812 Reduces Growth of LNCaP and 22Rv1 Cells in a Dose-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. In Silico Experiments
4.1.1. Homology Modelling of N-Myc-Max Heterodimer
4.1.2. Protein and Ligand Preparation
4.1.3. Molecular Docking
4.1.4. Molecular Dynamics Simulations
4.2. In Vitro Evaluation of Hit Compounds
4.2.1. eGFP Cellular Myc Transcription Assay
4.2.2. Cell Viability Assays
4.2.3. Aurora A Kinase Activity Assay
4.2.4. Biolayer Interferometry Assay
5. Conclusions
6. Patent
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AR | Androgen receptor |
| AURKA | Aurora A kinase |
| bHLHLZ | basic helix-loop-helix leucine zipper |
| BLI | bio-layer interferometry |
| CRPC | castration-resistant prostate cancer |
| DBD | DNA-binding domain |
| NEPC | neuroendocrine prostate cancer |
| PCa | prostate cancer |
| RMSD | Root-Mean Square Deviation |
| MD | Molecular dynamics |
Appendix A

References
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The Expanding World of N-MYC–Driven Tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebello, R.; Pearson, R.; Hannan, R.; Furic, L. Therapeutic Approaches Targeting MYC-Driven Prostate Cancer. Genes 2017, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attard, G.; Borre, M.; Gurney, H.; Loriot, Y.; Andresen-Daniil, C.; Kalleda, R.; Pham, T.; Taplin, M.-E.; on behalf of the PLATO Collaborators. Abiraterone Alone or in Combination with Enzalutamide in Metastatic Castration-Resistant Prostate Cancer with Rising Prostate-Specific Antigen During Enzalutamide Treatment. JCO 2018, 36, 2639–2646. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, D.J.; Annala, M.; Taavitsainen, S.; Finch, D.L.; Oja, C.; Vergidis, J.; Zulfiqar, M.; Sunderland, K.; Azad, A.A.; Kollmannsberger, C.K.; et al. Optimal sequencing of enzalutamide and abiraterone acetate plus prednisone in metastatic castration-resistant prostate cancer: A multicentre, randomised, open-label, phase 2, crossover trial. Lancet Oncol. 2019, 20, 1730–1739. [Google Scholar] [CrossRef]
- Beltran, H.; Hruszkewycz, A.; Scher, H.I.; Hildesheim, J.; Isaacs, J.; Yu, E.Y.; Kelly, K.; Lin, D.; Dicker, A.P.; Arnold, J.T.; et al. The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res. 2019, 25, 6916–6924. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, N.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun. 2018, 9, 4080. [Google Scholar] [CrossRef]
- Conteduca, V.; Oromendia, C.; Eng, K.W.; Bareja, R.; Sigouros, M.; Molina, A.; Faltas, B.M.; Sboner, A.; Mosquera, J.M.; Elemento, O.; et al. Clinical features of neuroendocrine prostate cancer. Eur. J. Cancer 2019, 121, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Investig. 2019, 130, 3924–3940. [Google Scholar] [CrossRef]
- Yin, Y.; Xu, L.; Chang, Y.; Zeng, T.; Chen, X.; Wang, A.; Groth, J.; Foo, W.-C.; Liang, C.; Hu, H.; et al. N-Myc promotes therapeutic resistance development of neuroendocrine prostate cancer by differentially regulating miR-421/ATM pathway. Mol. Cancer 2019, 18, 11. [Google Scholar] [CrossRef]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, A.; Gao, K.; Chu, L.; Zhang, R.; Yang, J.; Zheng, J. Aurora kinases: Novel therapy targets in cancers. Oncotarget 2017, 8, 23937–23954. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.C.Y.; Frolov, A.; Li, R.; Ayala, G.; Greenberg, N.M. Targeting Aurora Kinases for the Treatment of Prostate Cancer. Cancer Res. 2006, 66, 4996–5002. [Google Scholar] [CrossRef] [Green Version]
- Rong, R.; Jiang, L.Y.; Sheikh, M.S.; Huang, Y. Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene 2007, 26, 7700–7708. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.; Noble, M.; Wedge, S.R.; Robson, C.N.; Gaughan, L. Aurora A regulates expression of AR-V7 in models of castrate resistant prostate cancer. Sci. Rep. 2017, 7, 40957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nna, E.; Madukwe, J.; Egbujo, E.; Obiorah, C.; Okolie, C.; Echejoh, G.; Yahaya, A.; Adisa, J.; Uzoma, I. Gene expression of Aurora kinases in prostate cancer and nodular hyperplasia tissues. Med. Princ. Pract. 2013, 22, 138–143. [Google Scholar] [CrossRef]
- Nikonova, A.S.; Astsaturov, I.; Serebriiskii, I.G.; Dunbrack, R.L.; Golemis, E.A. Aurora A kinase (AURKA) in normal and pathological cell division. Cell. Mol. Life Sci. 2013, 70, 661–687. [Google Scholar] [CrossRef]
- Beltran, H.; Oromendia, C.; Danila, D.C.; Montgomery, B.; Hoimes, C.; Szmulewitz, R.Z.; Vaishampayan, U.; Armstrong, A.J.; Stein, M.; Pinski, J.; et al. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Wang, C.; He, B.; Yang, M.; Tong, M.; Long, Z.; Liu, B.; Peng, F.; Xu, L.; Zhang, Y.; et al. Aurora-A Kinase: A Potent Oncogene and Target for Cancer Therapy: AURORA-A KINASE IN CANCER. Med. Res. Rev. 2016, 36, 1036–1079. [Google Scholar] [CrossRef]
- Richards, M.W.; Burgess, S.G.; Poon, E.; Carstensen, A.; Eilers, M.; Chesler, L.; Bayliss, R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 13726–13731. [Google Scholar] [CrossRef] [Green Version]
- Kivinummi, K.; Urbanucci, A.; Leinonen, K.; Tammela, T.L.J.; Annala, M.; Isaacs, W.B.; Bova, G.S.; Nykter, M.; Visakorpi, T. The expression of AURKA is androgen regulated in castration-resistant prostate cancer. Sci. Rep. 2017, 7, 17978. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular Characterization of Neuroendocrine Prostate Cancer and Identification of New Drug Targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carabet, L.A.; Lallous, N.; Leblanc, E.; Ban, F.; Morin, H.; Lawn, S.; Ghaidi, F.; Lee, J.; Mills, I.G.; Gleave, M.E.; et al. Computer-aided drug discovery of Myc-Max inhibitors as potential therapeutics for prostate cancer. Eur. J. Med. Chem. 2018, 160, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafson, W.C.; Meyerowitz, J.G.; Nekritz, E.A.; Chen, J.; Benes, C.; Charron, E.; Simonds, E.F.; Seeger, R.; Matthay, K.K.; Hertz, N.T.; et al. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 2014, 26, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Hurlin, P.J. N-myc functions in transcription and development. Birth Defect Res. C 2005, 75, 340–352. [Google Scholar] [CrossRef]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Masso-Valles, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Sharma, N.; Giri, R. Therapeutic Interventions of Cancers Using Intrinsically Disordered Proteins as Drug Targets: C-Myc as Model System. Cancer Inform. 2017, 16. [Google Scholar] [CrossRef]
- Huang, M.-J.; Cheng, Y.; Liu, C.-R.; Lin, S.; Liu, H.E. A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle arrest, apoptosis, and myeloid differentiation of human acute myeloid leukemia. Exp. Hematol. 2006, 34, 1480–1489. [Google Scholar] [CrossRef]
- Yap, J.L.; Wang, H.; Hu, A.; Chauhan, J.; Jung, K.-Y.; Gharavi, R.B.; Prochownik, E.V.; Fletcher, S. Pharmacophore identification of c-Myc inhibitor 10074-G5. Bioorganic Med. Chem. Lett. 2013, 23, 370–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, J.R.; Garner, A.L.; Yu, J.; Ito, Y.; Sun, M.; Ueno, L.; Rhee, J.-K.; Baksh, M.M.; Stefan, E.; Hartl, M.; et al. Inhibitor of MYC identified in a Krohnke pyridine library. Proc. Natl. Acad. Sci. USA 2014, 111, 12556–12561. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chauhan, J.; Hu, A.; Pendleton, K.; Yap, J.L.; Sabato, P.E.; Jones, J.W.; Perri, M.; Yu, J.; Cione, E.; et al. Disruption of Myc-Max Heterodimerization with Improved Cell-Penetrating Analogs of the Small Molecule 10074-G5. Oncotarget 2013, 4, 936–947. [Google Scholar] [CrossRef]
- Kiessling, A.; Wiesinger, R.; Sperl, B.; Berg, T. Selective Inhibition of c-Myc/Max Dimerization by a Pyrazolo[1,5-a]pyrimidine. ChemMedChem 2007, 2, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.-C.; Kim, K.-T.; Seo, H.-H.; Shin, S.-P.; Ahn, K.-O.; Ji, M.-J.; Park, W.S.; Kim, I.-H.; Lee, S.-J.; Seo, H.K. Intravesical Instillation of c-MYC Inhibitor KSI-3716 Suppresses Orthotopic Bladder Tumor Growth. J. Urol. 2014, 191, 510–518. [Google Scholar] [CrossRef]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef] [Green Version]
- Carabet, L.A.; Rennie, P.S.; Cherkasov, A. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. Int. J. Mol. Sci. 2018, 20, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitfield, J.R.; Beaulieu, M.-E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.K.; Ahn, K.-O.; Jung, N.-R.; Shin, J.-S.; Park, W.S.; Lee, K.-H.; Lee, S.-J.; Jeong, K.-C. Antitumor activity of the c-Myc inhibitor KSI-3716 in gemcitabine-resistant bladder cancer. Oncotarget 2014, 5, 326–337. [Google Scholar] [CrossRef] [Green Version]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Mossé, Y.P.; Lipsitz, E.; Fox, E.; Teachey, D.T.; Maris, J.M.; Weigel, B.; Adamson, P.C.; Ingle, M.A.; Ahern, C.H.; Blaney, S.M. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: A Children’s Oncology Group Phase I Consortium study. Clin. Cancer Res. 2012, 18, 6058–6064. [Google Scholar] [CrossRef] [Green Version]
- Martens, S.; Goossens, V.; Devisscher, L.; Hofmans, S.; Claeys, P.; Vuylsteke, M.; Takahashi, N.; Augustyns, K.; Vandenabeele, P. RIPK1-dependent cell death: A novel target of the Aurora kinase inhibitor Tozasertib (VX-680). Cell Death Dis. 2018, 9, 211. [Google Scholar] [CrossRef] [Green Version]
- Bavetsias, V.; Linardopoulos, S. Aurora Kinase Inhibitors: Current Status and Outlook. Front. Oncol. 2015, 5, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisa, A.C.; Bhatt, H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017, 140, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Nikhil, K.; Raza, A.; Haymour, H.S.; Flueckiger, B.V.; Chu, J.; Shah, K. Aurora Kinase A-YBX1 Synergy Fuels Aggressive Oncogenic Phenotypes and Chemoresistance in Castration-Resistant Prostate Cancer. Cancers 2020, 12, 660. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.K.; Burley, S.K. X-Ray Structures of Myc-Max and Mad-Max Recognizing DNA. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Boike, L.; Cioffi, A.G.; Majewski, F.C.; Co, J.; Henning, N.J.; Jones, M.D.; Liu, G.; McKenna, J.M.; Tallarico, J.A.; Schirle, M.; et al. Discovery of a Functional Covalent Ligand Targeting an Intrinsically Disordered Cysteine within MYC. Cell Chem. Biol. 2020, 27, 1–10. [Google Scholar] [CrossRef]
- Kummar, S.; Gutierrez, M.; Doroshow, J.H.; Murgo, A.J. Drug development in oncology: Classical cytotoxics and molecularly targeted agents. Br. J. Clin. Pharmacol. 2006, 62, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Chemical Computing Group ULC. Molecular Operating Environment (MOE); 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2018. [Google Scholar]
- Nayeem, A.; Sitkoff, D.; Krystek, S. A comparative study of available software for high-accuracy homology modeling: From sequence alignments to structural models. Protein Sci. 2006, 15, 808–824. [Google Scholar] [CrossRef] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.J.; Sacerdoti, F.D.; Salmon, J.K.; Shan, Y.; Shaw, D.E.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; et al. Molecular Dynamics—Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing—SC ’06, Tampa, FL, USA, 11–17 November 2006; ACM Press: New York, NY, USA, 2006; p. 84. [Google Scholar]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ton, A.-T.; Singh, K.; Morin, H.; Ban, F.; Leblanc, E.; Lee, J.; Lallous, N.; Cherkasov, A. Dual-Inhibitors of N-Myc and AURKA as Potential Therapy for Neuroendocrine Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 8277. https://doi.org/10.3390/ijms21218277
Ton A-T, Singh K, Morin H, Ban F, Leblanc E, Lee J, Lallous N, Cherkasov A. Dual-Inhibitors of N-Myc and AURKA as Potential Therapy for Neuroendocrine Prostate Cancer. International Journal of Molecular Sciences. 2020; 21(21):8277. https://doi.org/10.3390/ijms21218277
Chicago/Turabian StyleTon, Anh-Tien, Kriti Singh, Hélène Morin, Fuqiang Ban, Eric Leblanc, Joseph Lee, Nada Lallous, and Artem Cherkasov. 2020. "Dual-Inhibitors of N-Myc and AURKA as Potential Therapy for Neuroendocrine Prostate Cancer" International Journal of Molecular Sciences 21, no. 21: 8277. https://doi.org/10.3390/ijms21218277