Tandem Electrospray Mass Spectrometry of Cyclic N-Substituted Oligo-β-(1→6)-D-glucosamines


Abstract
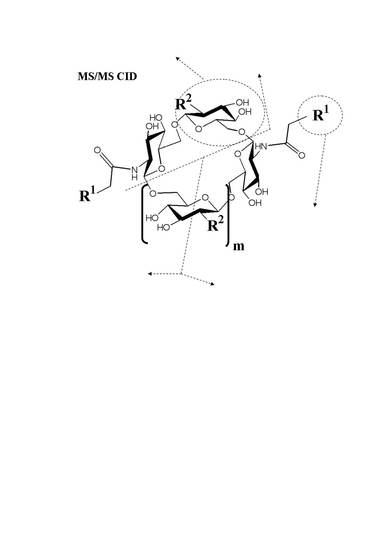
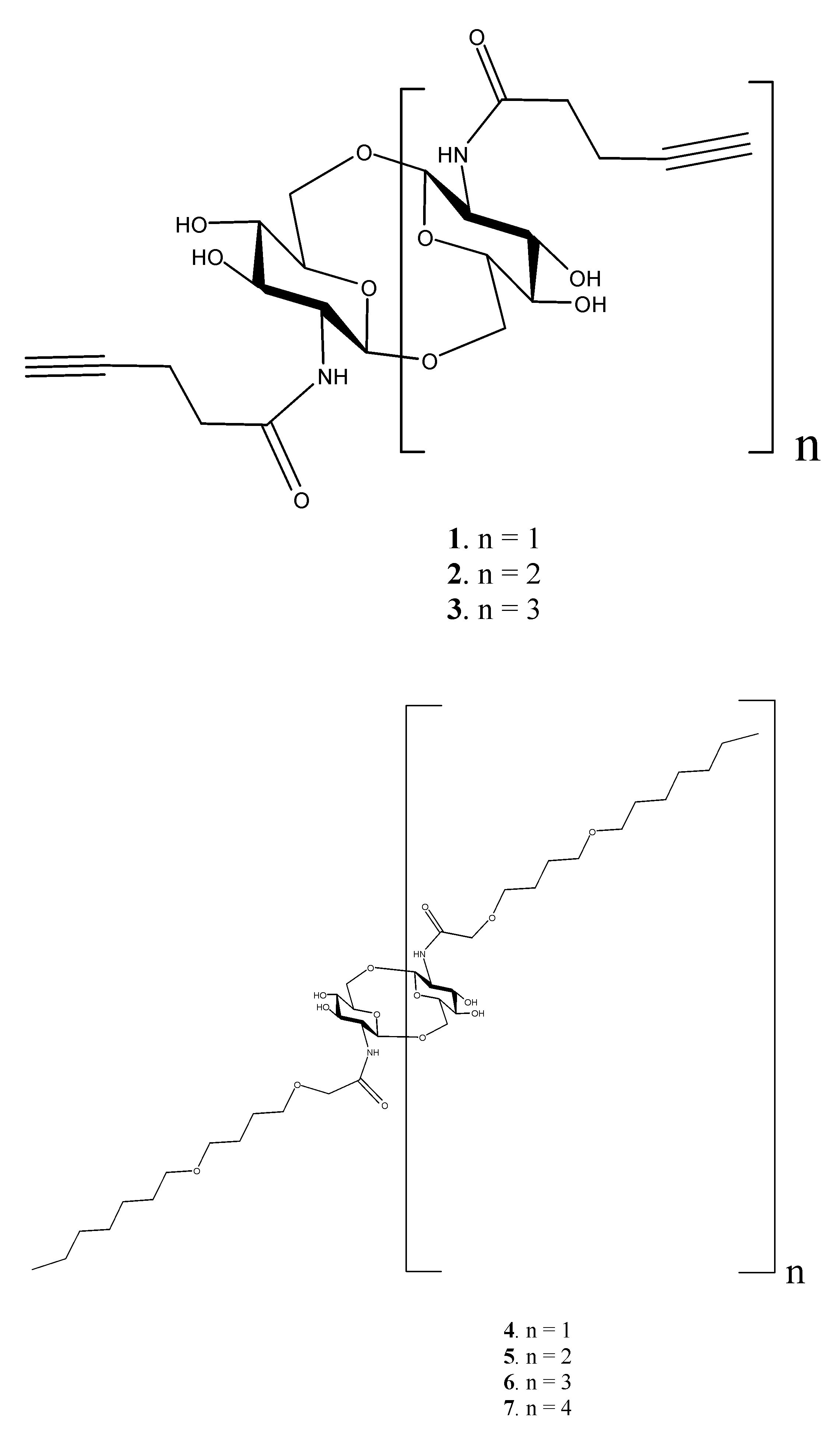
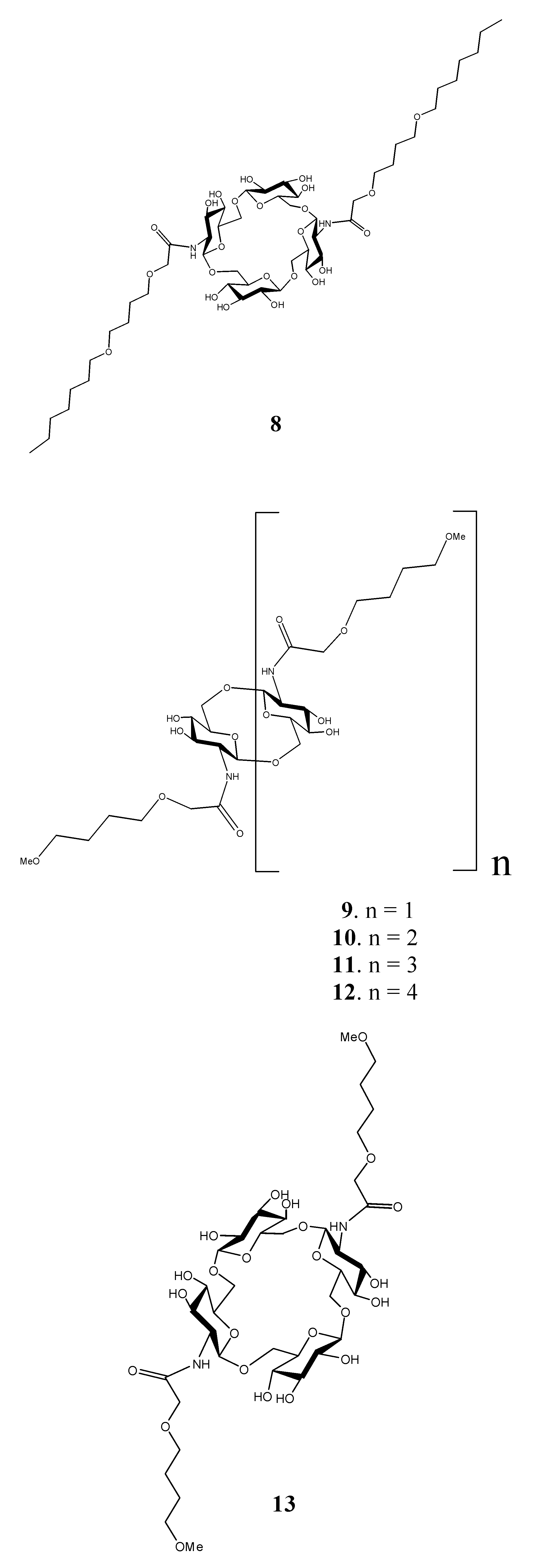
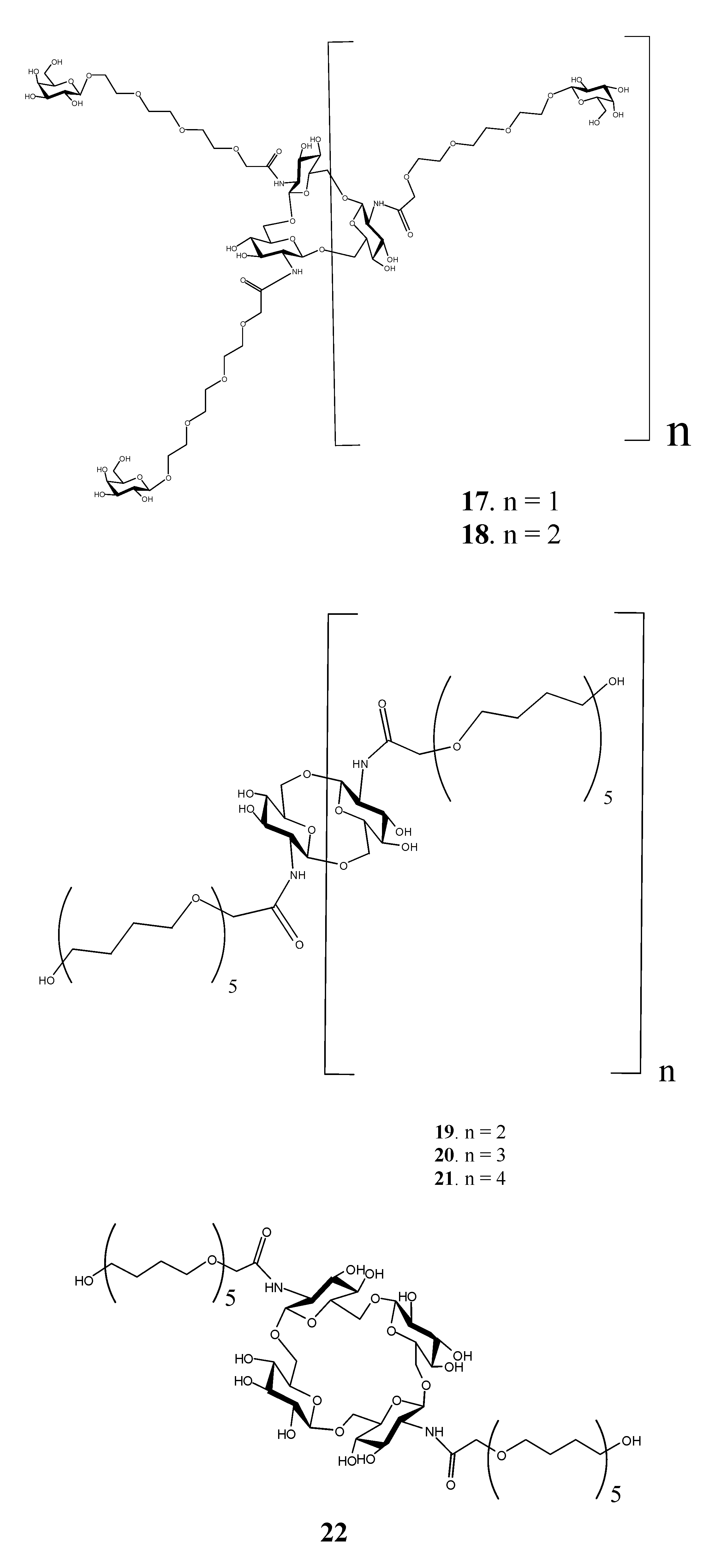
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
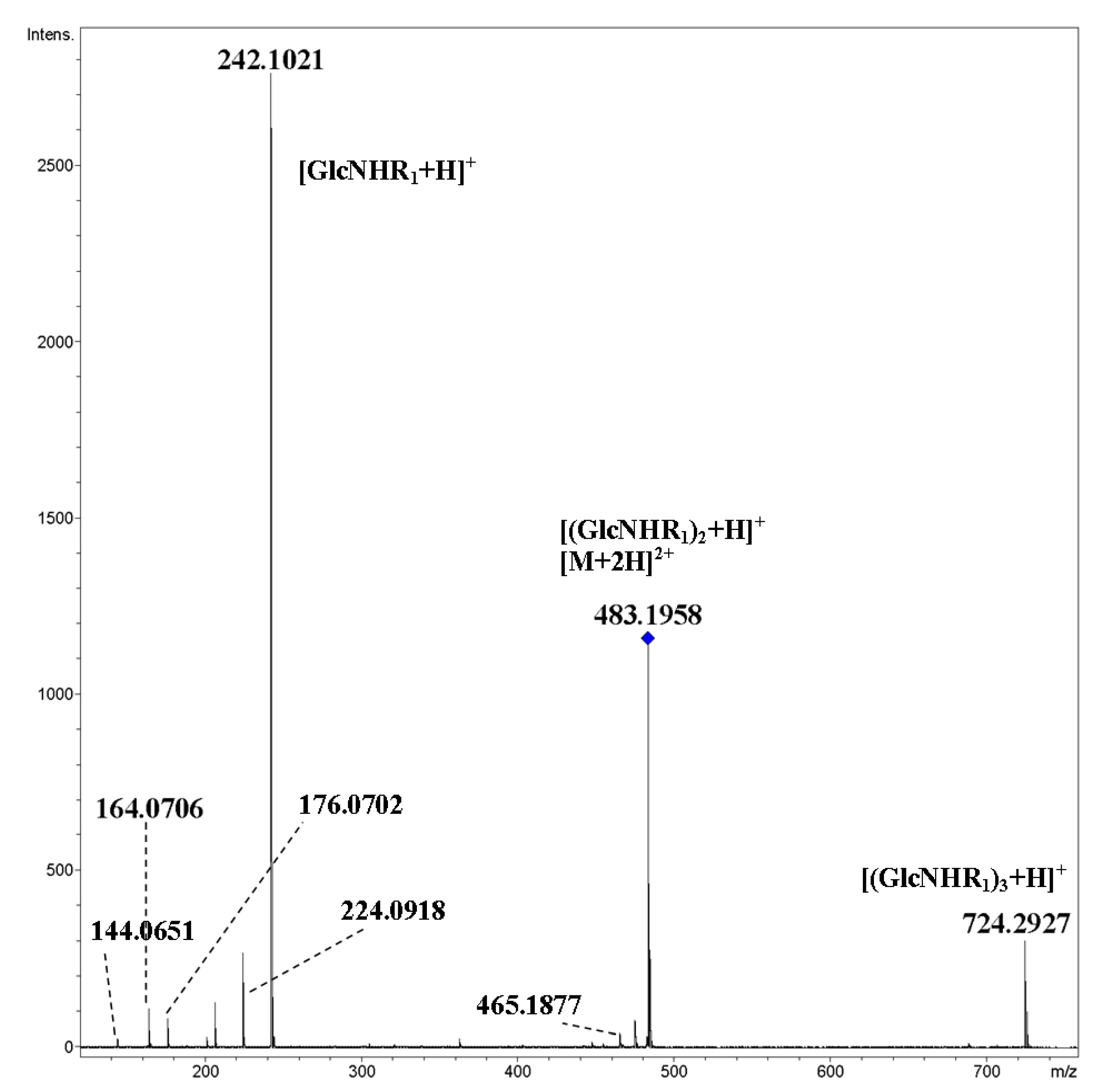
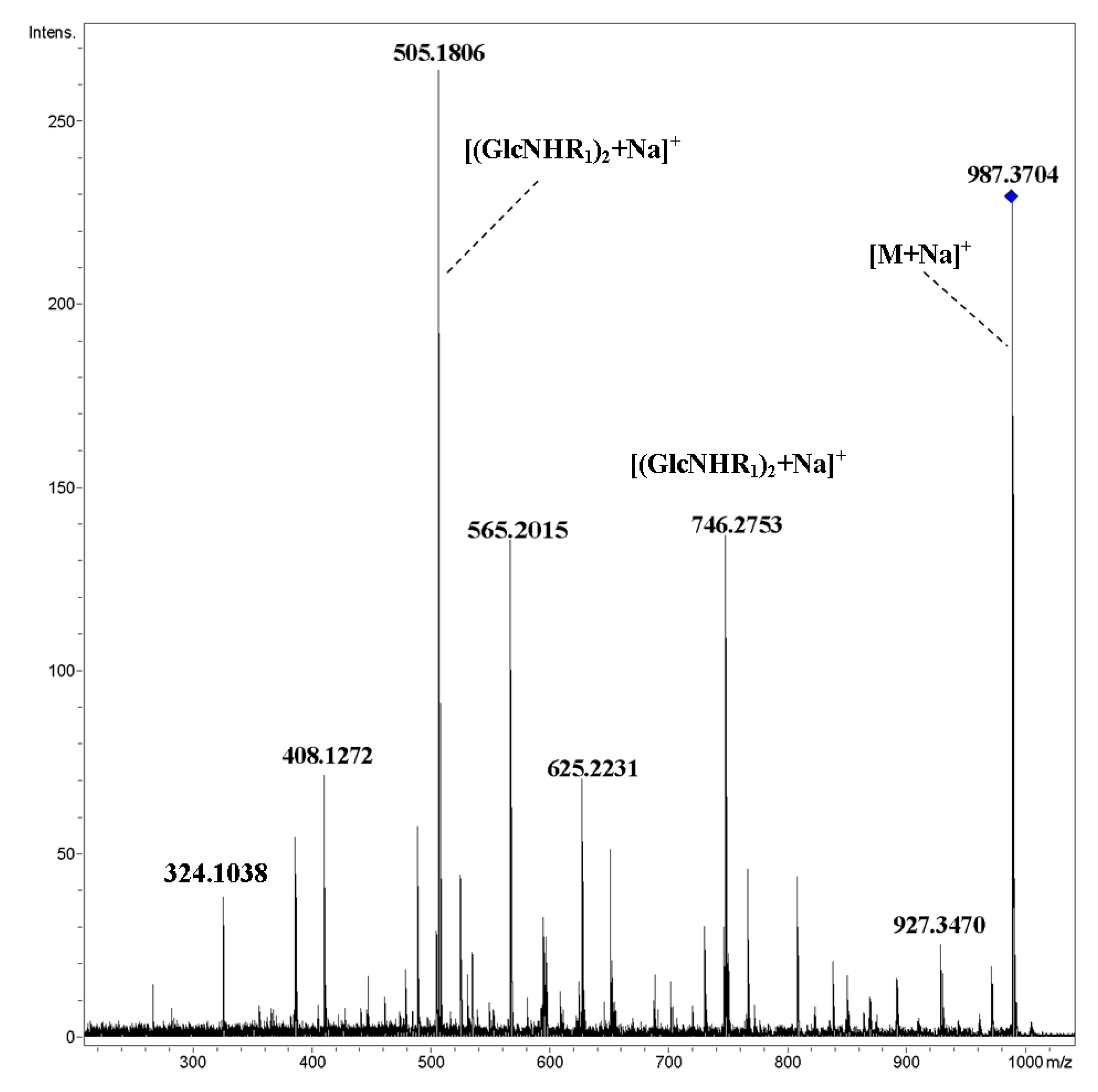
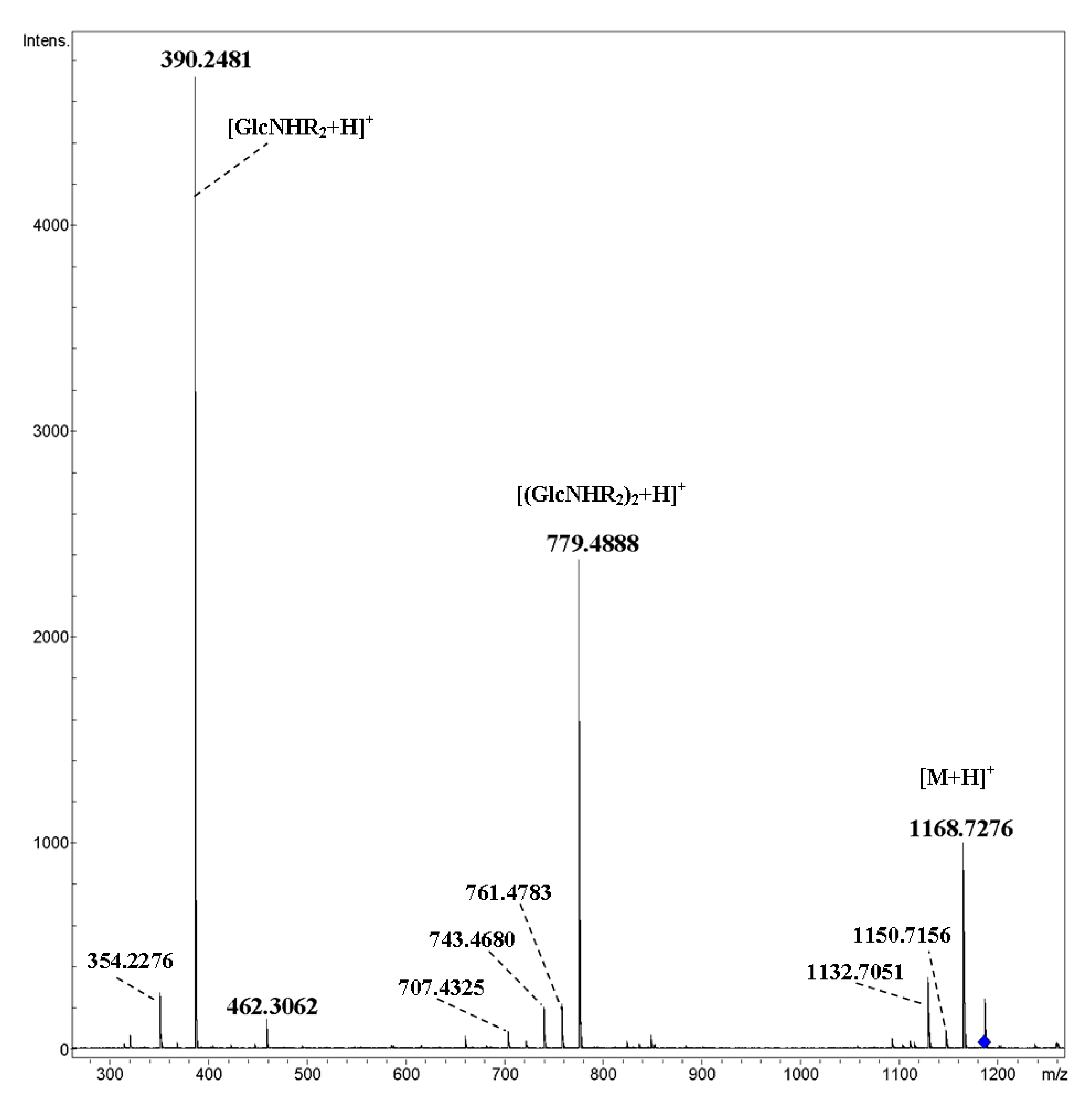
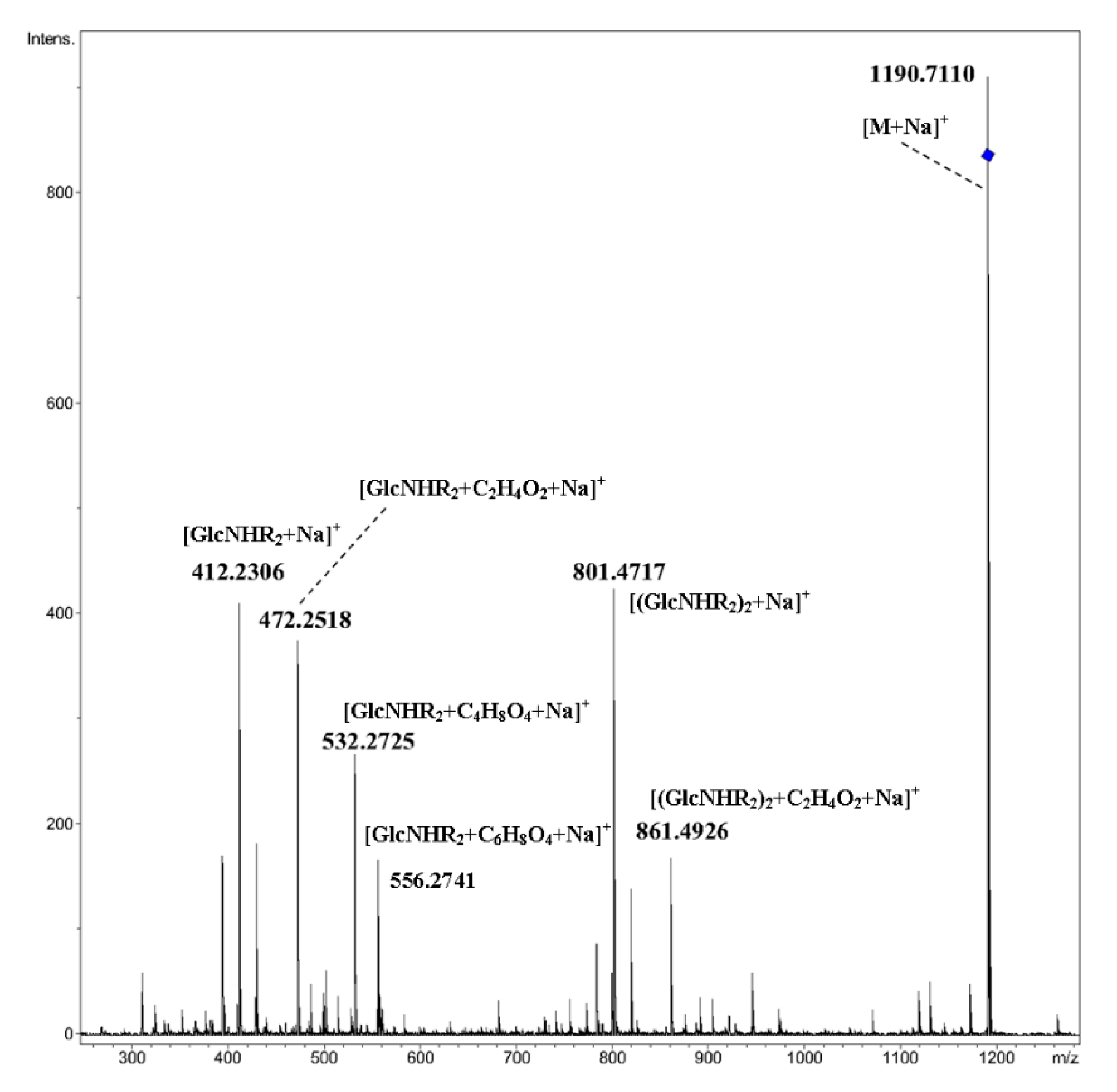
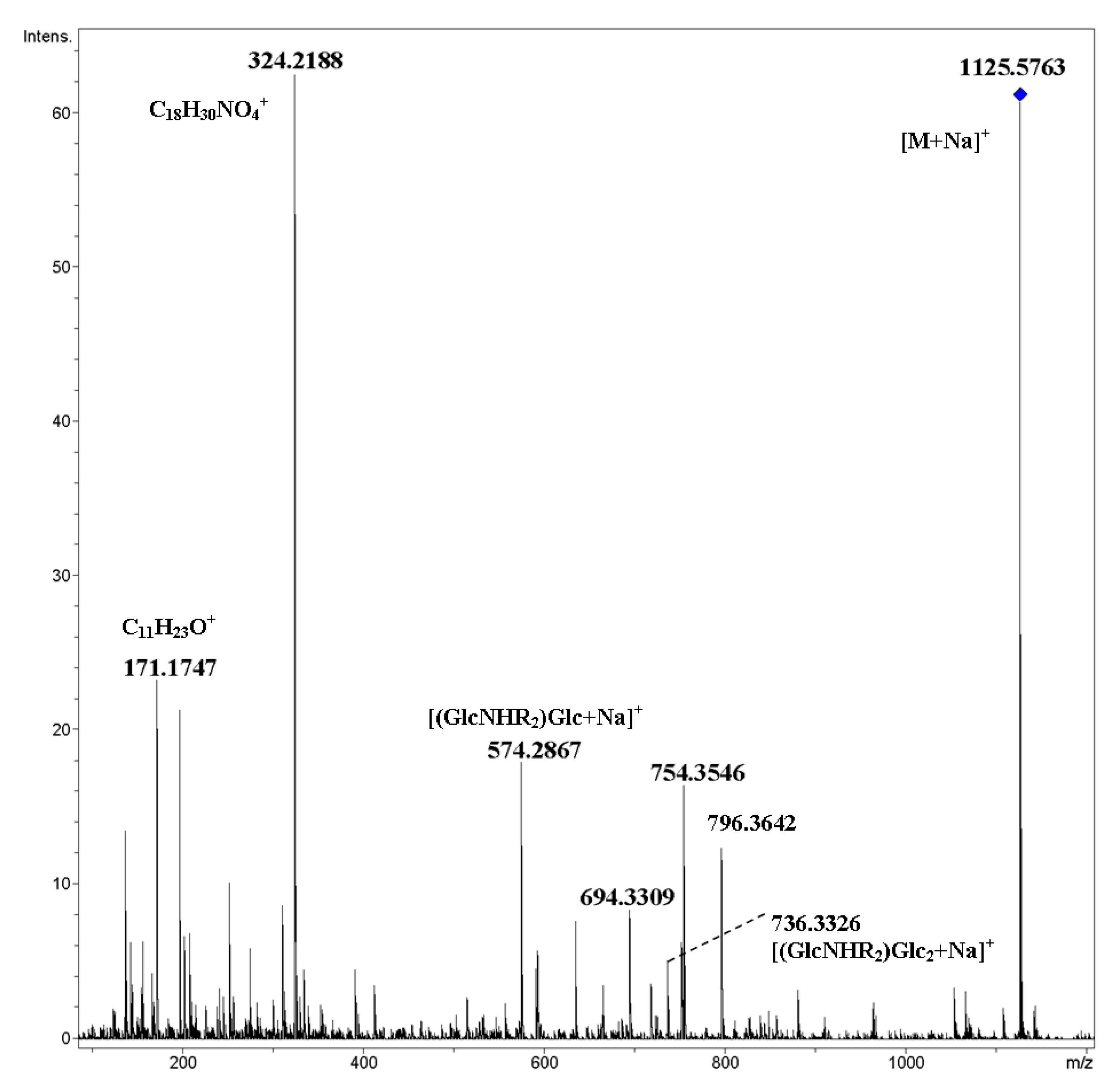
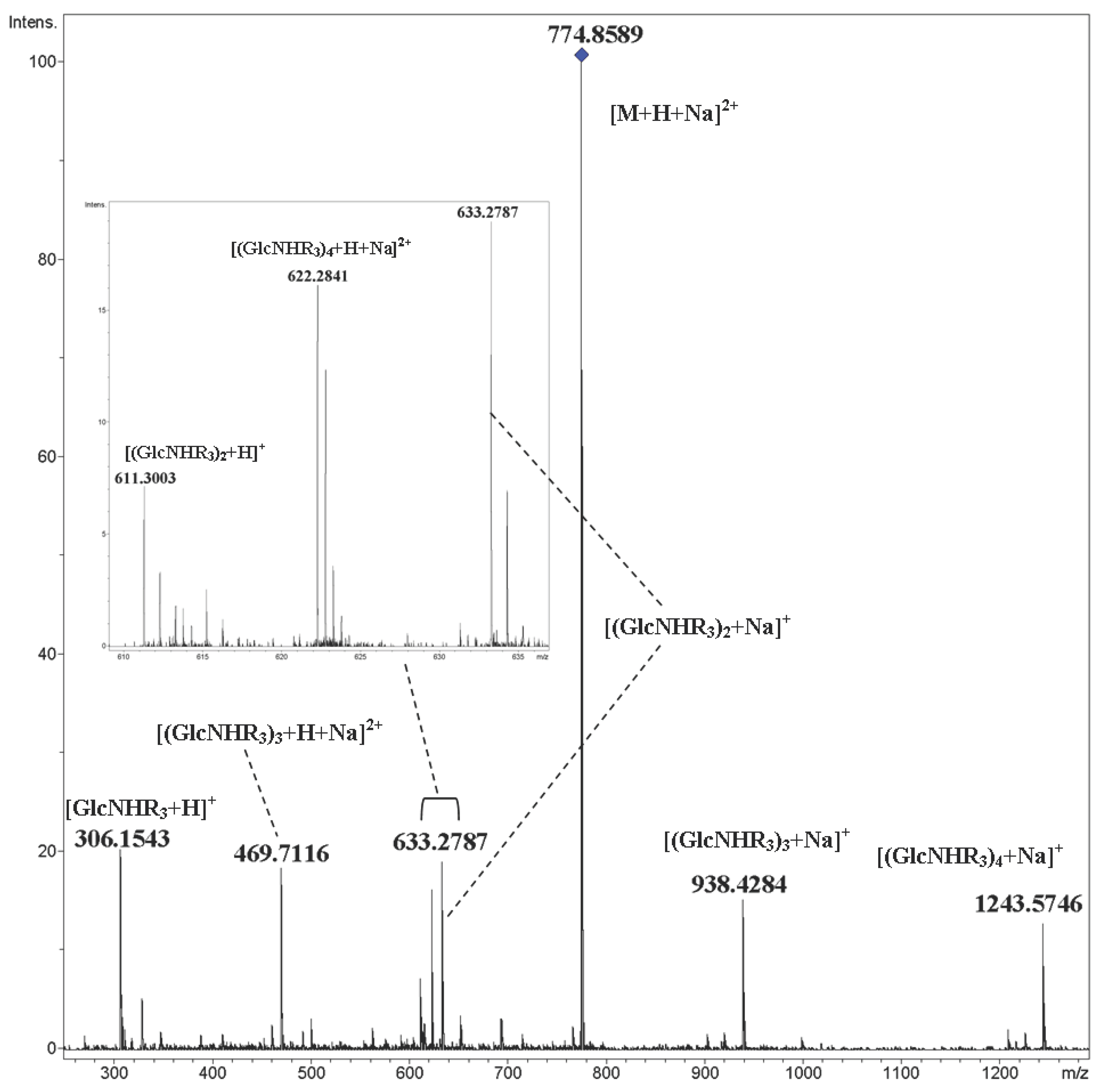
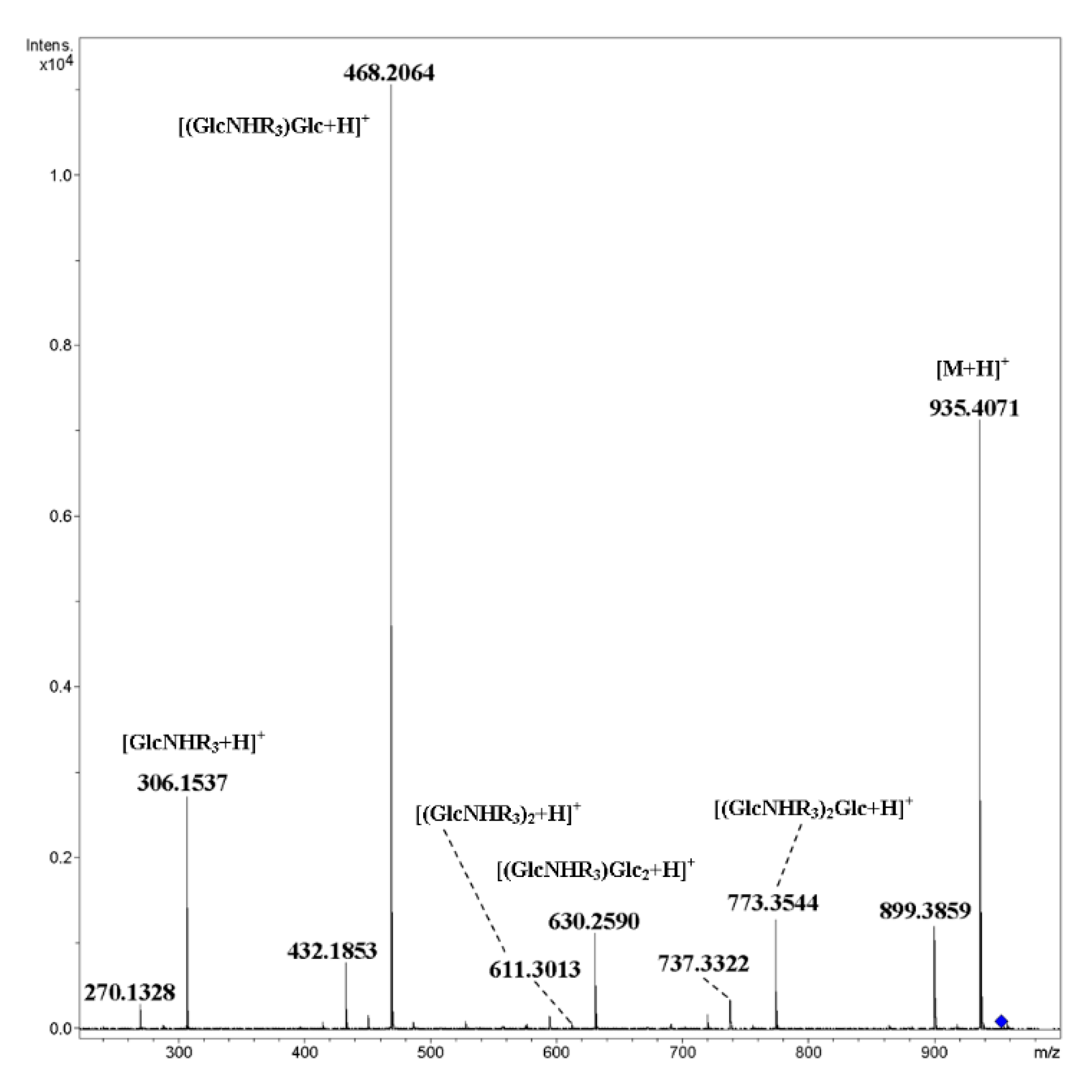
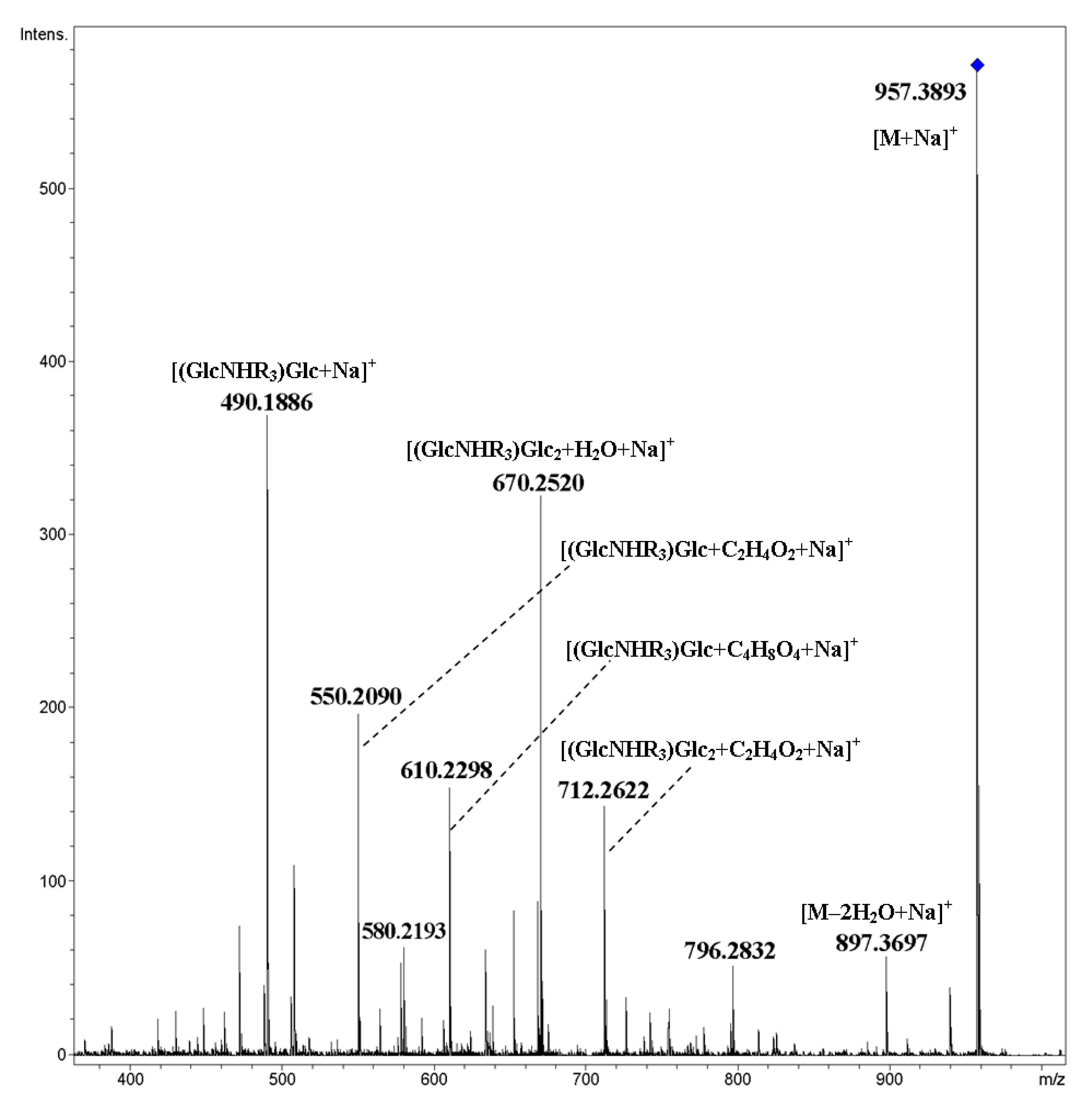
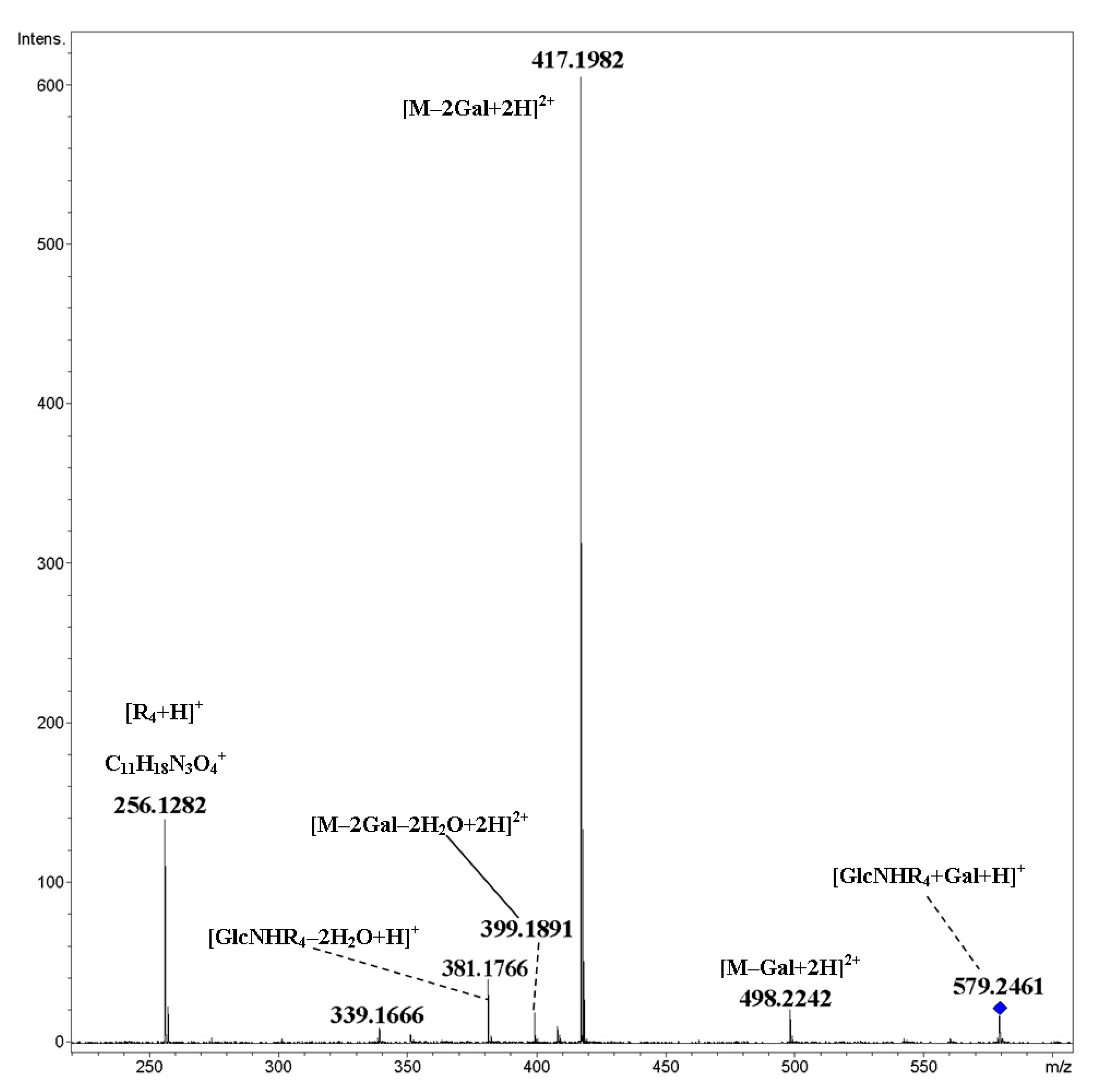
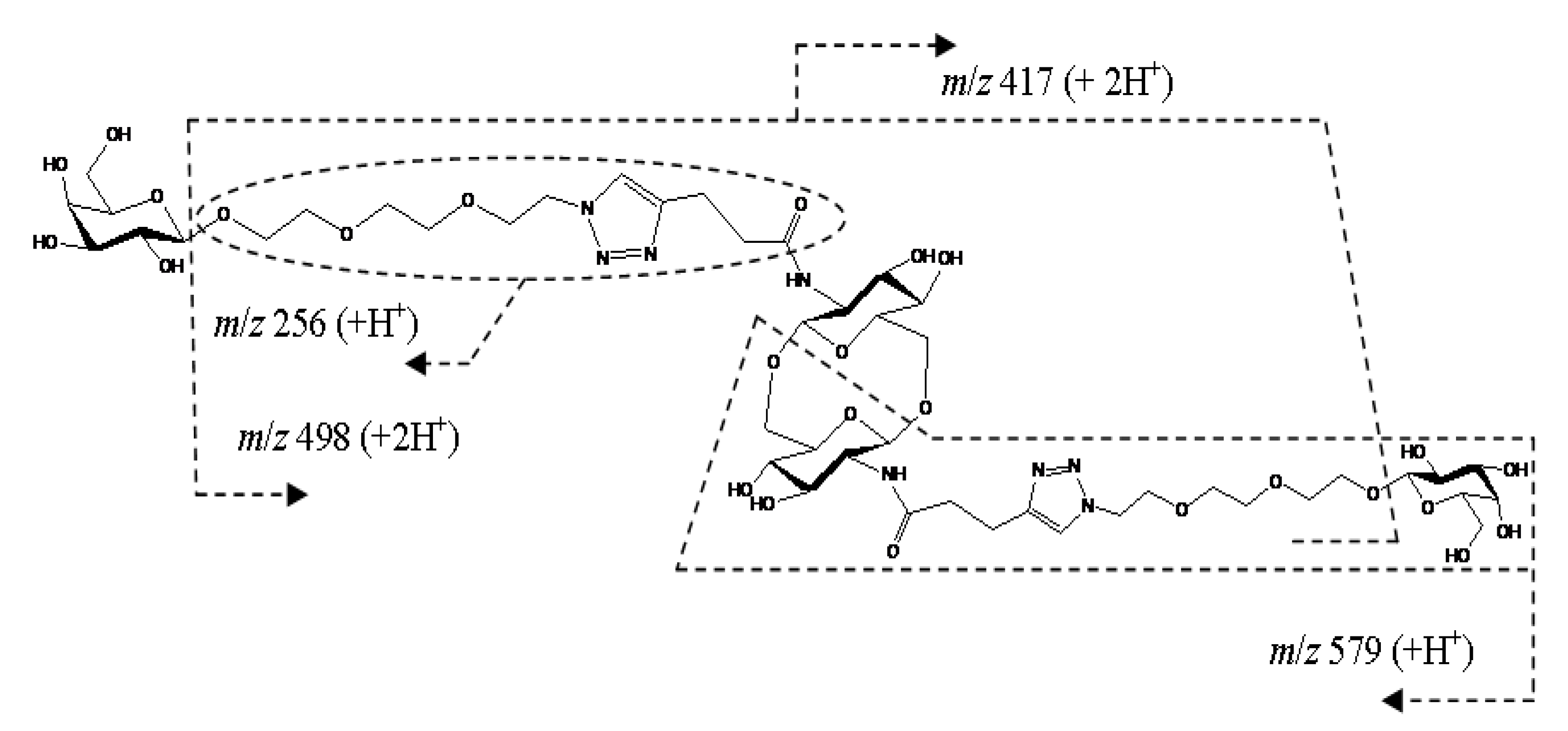
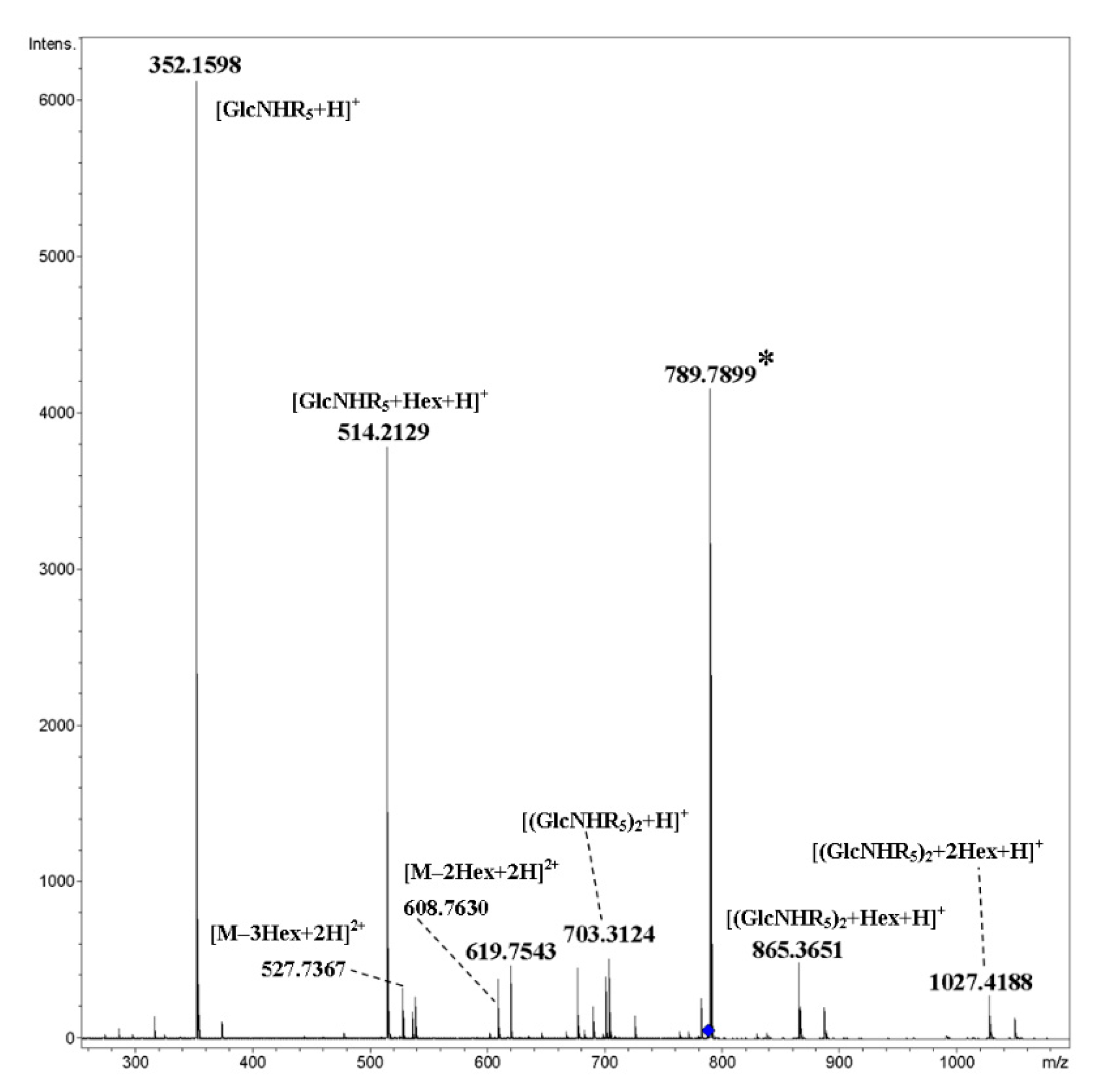
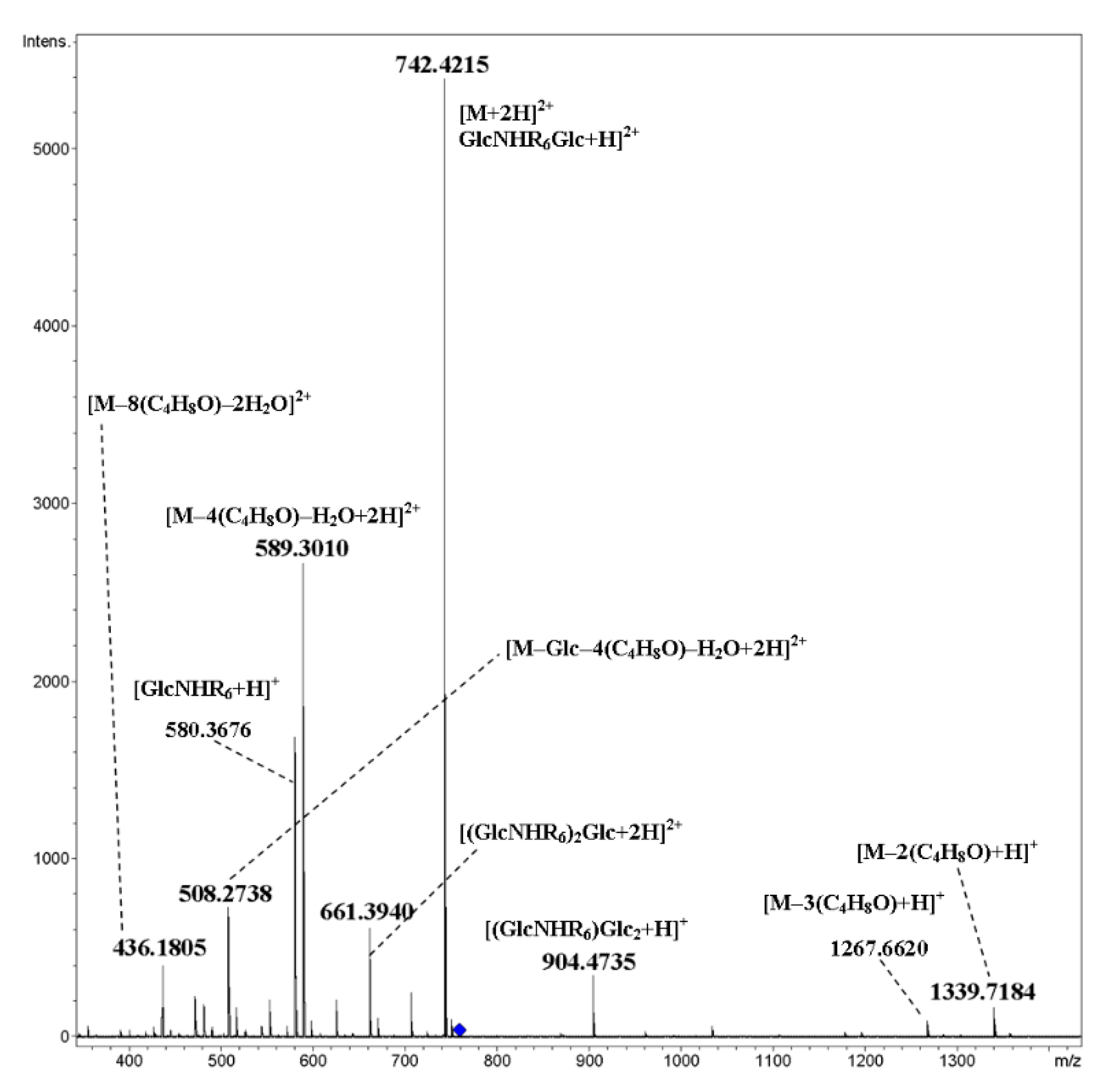
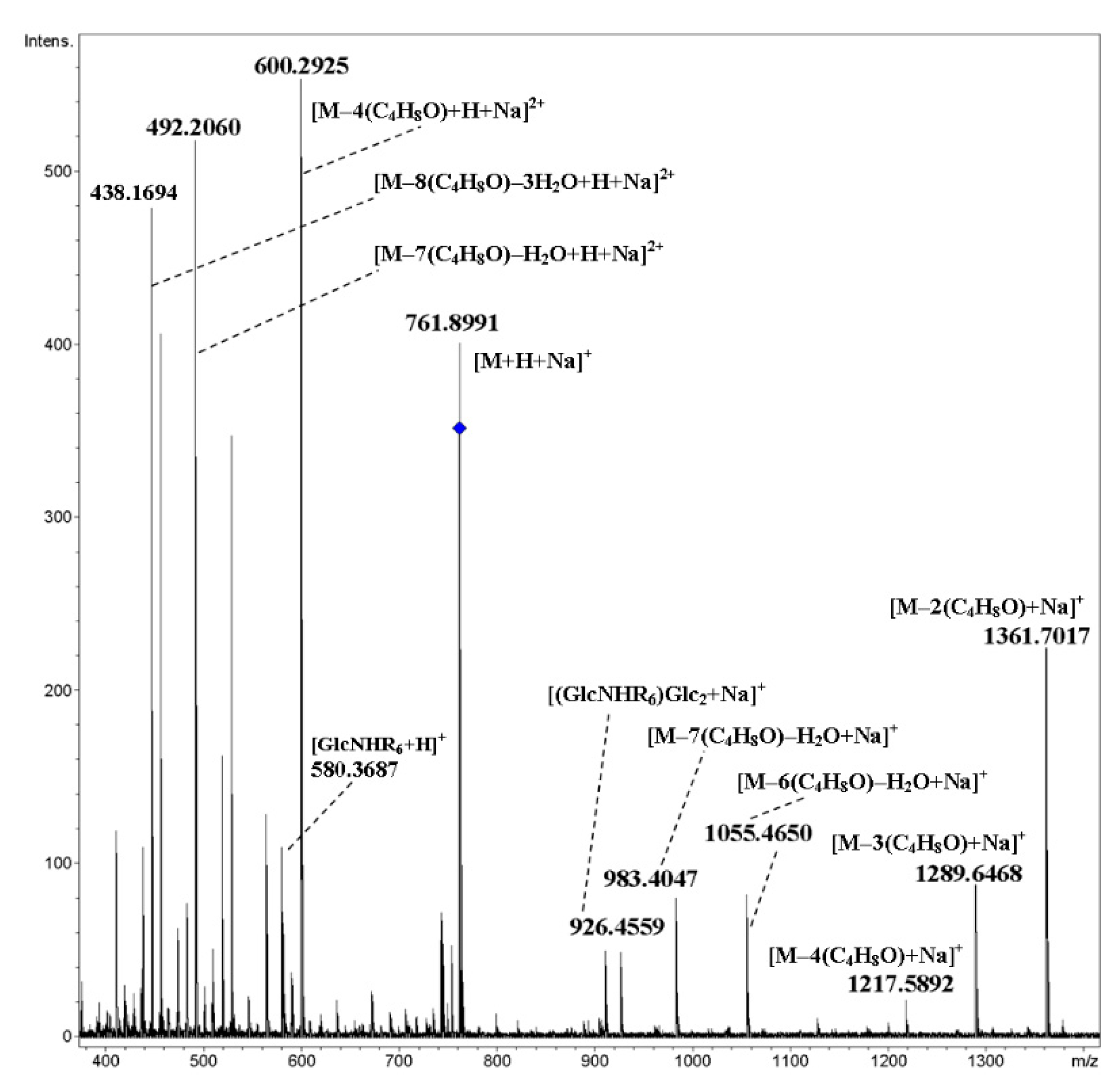
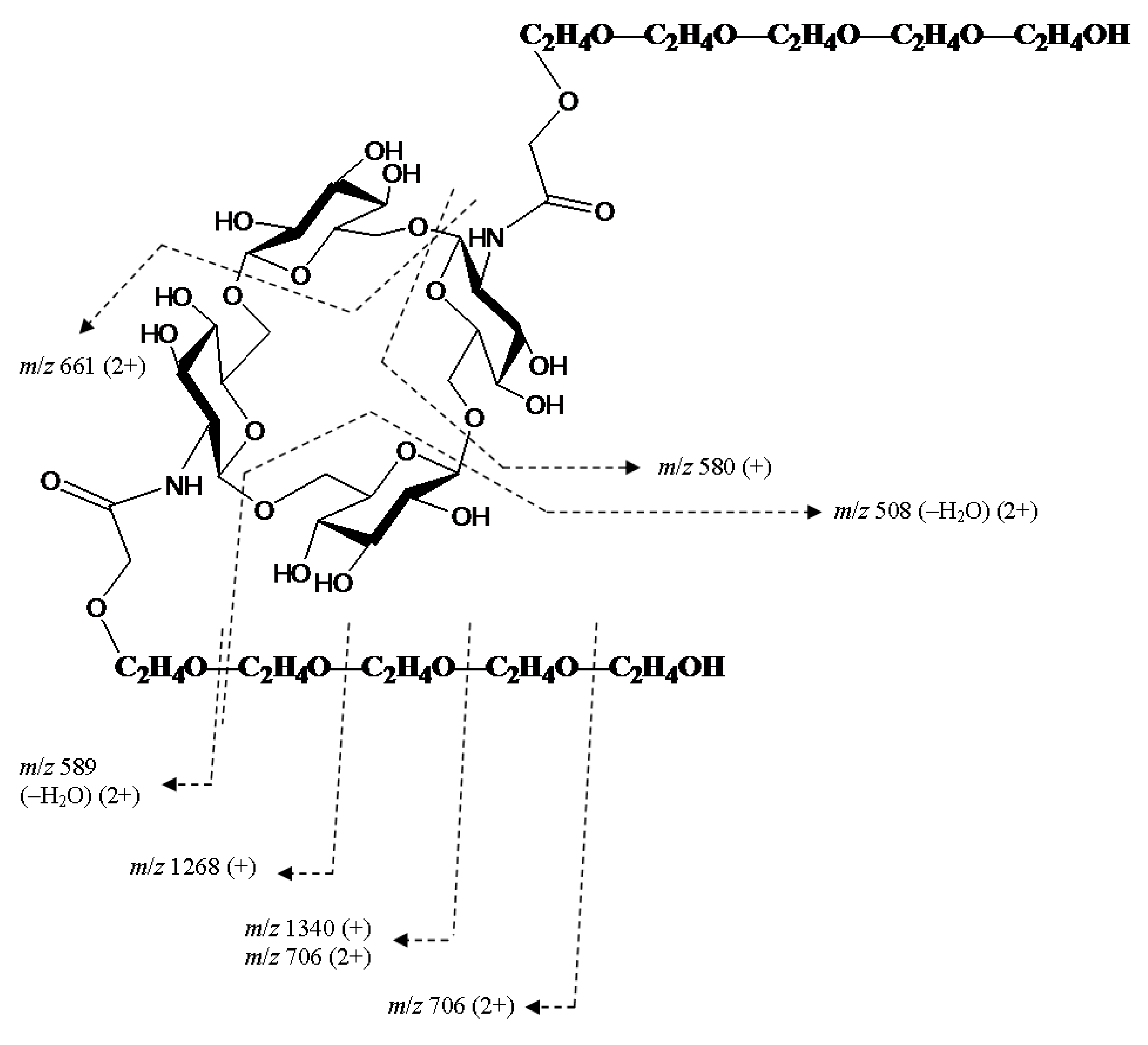
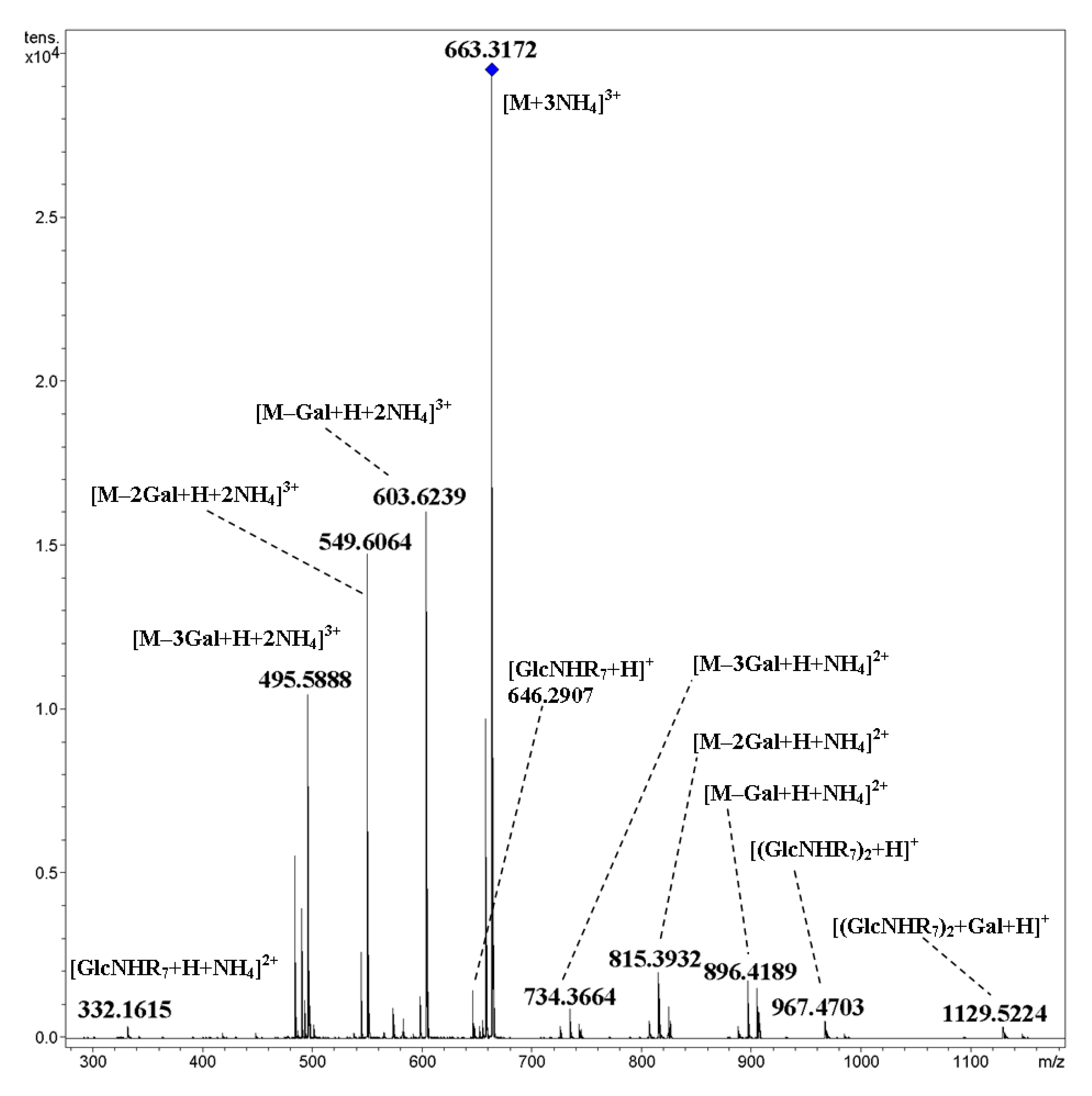
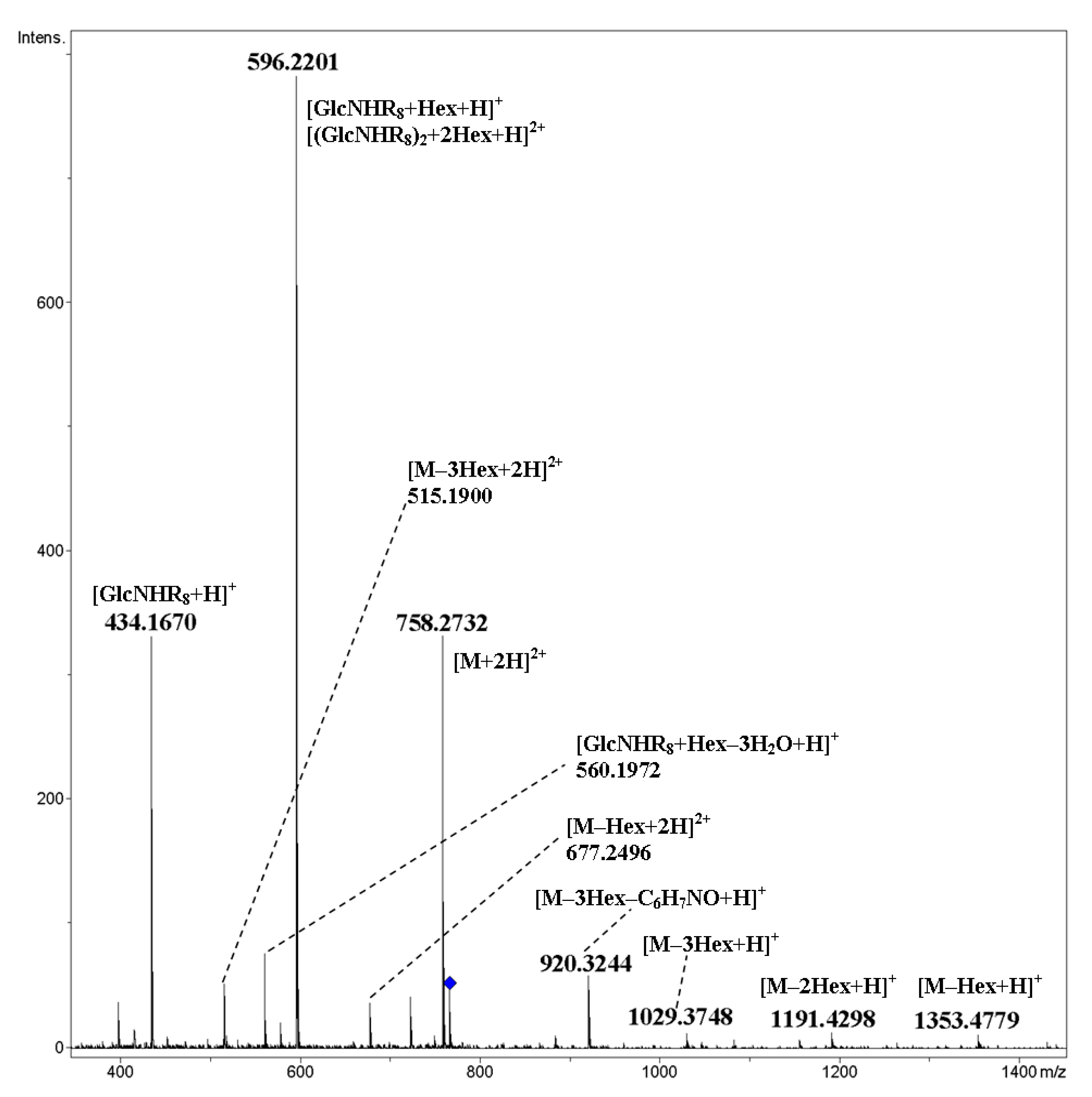
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CID | Collision-induced dissociation (decay) |
| Ea | Activation (collision) energy |
| ESI | Electrospray ionization |
| eV | Electron volt |
| Gal | Galactose |
| Glc | Glucose |
| GlcN | Glucosamine |
| GlcNHR | N-substituted glucosamine |
| Hex | Hexose |
| HRMS | High-resolution mass spectrometry |
| IRL | Internal residue loss |
| M | Molecule, molecular mass (in Da) |
| min | Minute |
| mL | Milliliter |
| [M+H]+ | Protonated molecule |
| [M+Met]+ | Metallated molecule |
| [M+NH4]+ | Ammoniated molecule |
| [M+Na]+ | Sodiated molecule |
| MS | Mass spectrometry, mass spectrum |
| MS/MS | Tandem mass spectrometry (2nd order) |
| m/z | Mass to charge ratio |
| QqToF | Quadrupole time-of-flight mass hybrid spectrometer |
| V | Volt |
| µL | Microliter |
References
- Crini, G. Review: A history of cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar] [CrossRef]
- Gening, M.L.; Tsvetkov Yu, E.; Pier, G.B.; Nifantiev, N.E. Investigation of the Reaction of Terminated Oligomerization in the Synthesis of oligo-β(1→6)-oligoglucosamines. Russ. J. Bioorg. Chem. 2006, 32, 389–399. [Google Scholar] [CrossRef]
- Gening, M.L.; Titov, D.V.; Grachev, A.A.; Gerbst, A.G.; Yudina, O.N.; Shashkov, A.S.; Chizhov, A.O.; Tsvetkov, Y.E.; Nifantiev, N.E. Synthesis, NMR, and conformational studies of cyclic oligo-(1→6)-β-D-glucosamines. Eur. J. Org. Chem. 2010, 2465–2475. [Google Scholar] [CrossRef]
- Gening, M.L.; Tsvetkov, Y.E.; Pier, G.B.; Nifantiev, N.E. Synthesis of oligo-β-(1→6)-glucosamines corresponding to the fragments of the surface polysaccharide of Staphylococcus aureus. Carbohydr. Res. 2007, 342, 567–575. [Google Scholar] [CrossRef]
- Titov, D.V.; Gening, M.L.; Gerbst, A.G.; Chizhov, A.O.; Tsvetkov, Y.E.; Nifantiev, N.E. Stereochemistry of intramolecular cyclization of tetra-β-(1→6)-D-glucosamines and related tetrasaccharides: The role of conformational stereocontrol and the neighboring group participation. Carbohydr. Res. 2013, 381, 161–178. [Google Scholar] [CrossRef]
- Titov, D.V.; Gening, M.L.; Tsvetkov, Y.E.; Nifantiev, N.E. Conjugates of cyclooligosaccharide scaffolds and carbohydrate ligands: Methods for synthesis and the interaction with lectins. Russ. J. Bioorg. Chem. 2013, 39, 451–481. [Google Scholar] [CrossRef]
- Grachev, A.A.; Gerbst, A.G.; Gening, M.L.; Titov, D.V.; Yudina, O.N.; Tsvetkov, Y.E.; Shashkov, A.S.; Pier, G.B.; Nifantiev, N.E. NMR and conformational studies of linear and cyclic oligo-(1→6)-β-D-glucosamines. Carbohydr. Res. 2011, 346, 2499–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, T.; Roy, A.; Gening, M.L.; Titov, D.V.; Gerbst, A.G.; Tsvetkov, Y.E.; Nifantiev, N.E.; Talukdar, P. Cyclo-oligo-(1→6)-β-D-glucosamine based artificial channels for tunable transmembrane ion transport. Chem. Commun. 2014, 50, 5514–5516. [Google Scholar] [CrossRef]
- Gening, M.L.; Tsvetkov, Y.E.; Titov, D.V.; Gerbst, A.G.; Yudina, O.N.; Grachev, A.A.; Shashkov, A.S.; Vidal, S.; Imberty, A.; Saha, T.; et al. Linear and cyclic oligo-β-(1→6)-D-glucosamines: Synthesis, conformations, and applications for design of a vaccine and oligodentate glycoconjugates. Pure Appl. Chem. 2013, 85, 1879–1892. [Google Scholar] [CrossRef] [Green Version]
- Gening, M.L.; Titov, D.V.; Cecioni, S.; Audfray, A.; Gerbst, A.G.; Tsvetkov, Y.E.; Krylov, V.B.; Imberty, A.; Nifantiev, N.E.; Vidal, S. Synthesis of multivalent carbohydrate-centered glycoclusters as nanomolar ligands of the bacterial lectin LecA from Pseudomonas aeruginosa. Chem. Eur. J. 2013, 19, 9272–9285. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Saha, T.; Gening, M.L.; Titov, D.V.; Gerbst, A.G.; Tsvetkov, Y.E.; Nifantiev, N.E.; Talukdar, P. Trimodal control of ion-transport activity of cyclo-oligo-(1→6)-β-D-glucosamine-based artificial ion-transport systems. Chem. Eur. J. 2015, 21, 17445–17452. [Google Scholar] [CrossRef]
- Chizhov, A.O.; Tsvetkov, Y.E.; Nifantiev, N.E. Gas-phase fragmentation of cyclic oligosaccharides in tandem mass spectrometry. Molecules 2019, 24, 2226. [Google Scholar] [CrossRef] [Green Version]
- Chizhov, A.O.; Gening, M.L.; Pinsker, O.A.; Yudina, O.N.; Tsvetkov, Y.E.; Nifantiev, N.E. Gas-phase fragmentation studies of cyclic oligo-β-(1→6)-D-glucosamines by electrospray ionization mass spectrometry using a hybrid high-resolution mass spectrometer. Russ. Chem. Bull. 2018, 67, 144–149. [Google Scholar] [CrossRef]
- Chizhov, A.O.; Gening, M.L.; Pinsker, O.A.; Tsvetkov, Y.E.; Nifantiev, N.E. Isomeric effects in collisionally-induced dissociation of β-(1→6)-linked cyclic tetrasaccharides of Glcp2GlcpN2. J. Anal. Chem. 2019, 74, 1320–1324. [Google Scholar] [CrossRef]
- Zaia, J. Mass spectrometry of oligosaccharides. Mass Spectrom. Rev. 2004, 23, 161–227. [Google Scholar] [CrossRef]
- Dongre, A.N.; Jones, J.N.; Somogyi, A.; Wysocki, V.H. Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: Evidence for the mobile proton model. J. Am. Chem. Soc. 1996, 118, 8365–8374. [Google Scholar] [CrossRef]
- Jacquet, R.; Elfakir, C.; Lafosse, M. Characterization of new methylated beta-cyclodextrin with low degree of substitution by electrospray ionization mass spectrometry and liquid chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 3097–3102. [Google Scholar] [CrossRef]
- Domon, B.; Costello, C.E. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 1988, 5, 397–409. [Google Scholar] [CrossRef]
- Wuhrer, M.; Deelder, A.M.; van der Burgt, Y.E.M. Mass spectrometric glycan rearrangements. Mass Spectrom. Rev. 2011, 30, 664–680. [Google Scholar] [CrossRef]
- Thirumrugan, P.; Matosiuk, D.; Jozwiak, K. Click chemistry for drug development add diverse chemical biology applications. Chem. Rev. 2013, 113, 4905–4979. [Google Scholar] [CrossRef] [PubMed]
- Chizhov, A.O.; Sukhova, E.V.; Khatuntseva, E.A.; Tsvetkov, Y.E.; Gening, M.L.; Nifantiev, N.E. High-resolution electrospray mass spectra of hexaethylene glycol connected biotinylated HNK-1 antigenic trisaccharide molecular probe and its non-sulfated analogue. Carbohydr. Res. 2015, 417, 15–18. [Google Scholar] [CrossRef]
- Tsedilin, A.M.; Fakhrutdinov, A.N.; Eremin, D.B.; Zalesskiy, S.S.; Chizhov, A.O.; Kolotyrkina, N.G.; Ananikov, V.P. How sensitive and accurate are routine NMR and MS measurements? Mendeleev Commun. 2015, 25, 454–456. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chizhov, A.O.; Gening, M.L.; Tsvetkov, Y.E.; Nifantiev, N.E. Tandem Electrospray Mass Spectrometry of Cyclic N-Substituted Oligo-β-(1→6)-D-glucosamines. Int. J. Mol. Sci. 2020, 21, 8284. https://doi.org/10.3390/ijms21218284
Chizhov AO, Gening ML, Tsvetkov YE, Nifantiev NE. Tandem Electrospray Mass Spectrometry of Cyclic N-Substituted Oligo-β-(1→6)-D-glucosamines. International Journal of Molecular Sciences. 2020; 21(21):8284. https://doi.org/10.3390/ijms21218284
Chicago/Turabian StyleChizhov, Alexander O., Marina L. Gening, Yury E. Tsvetkov, and Nikolay E. Nifantiev. 2020. "Tandem Electrospray Mass Spectrometry of Cyclic N-Substituted Oligo-β-(1→6)-D-glucosamines" International Journal of Molecular Sciences 21, no. 21: 8284. https://doi.org/10.3390/ijms21218284