Regulation of Osteoclast Differentiation at Multiple Stages by Protein Kinase D Family Kinases

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
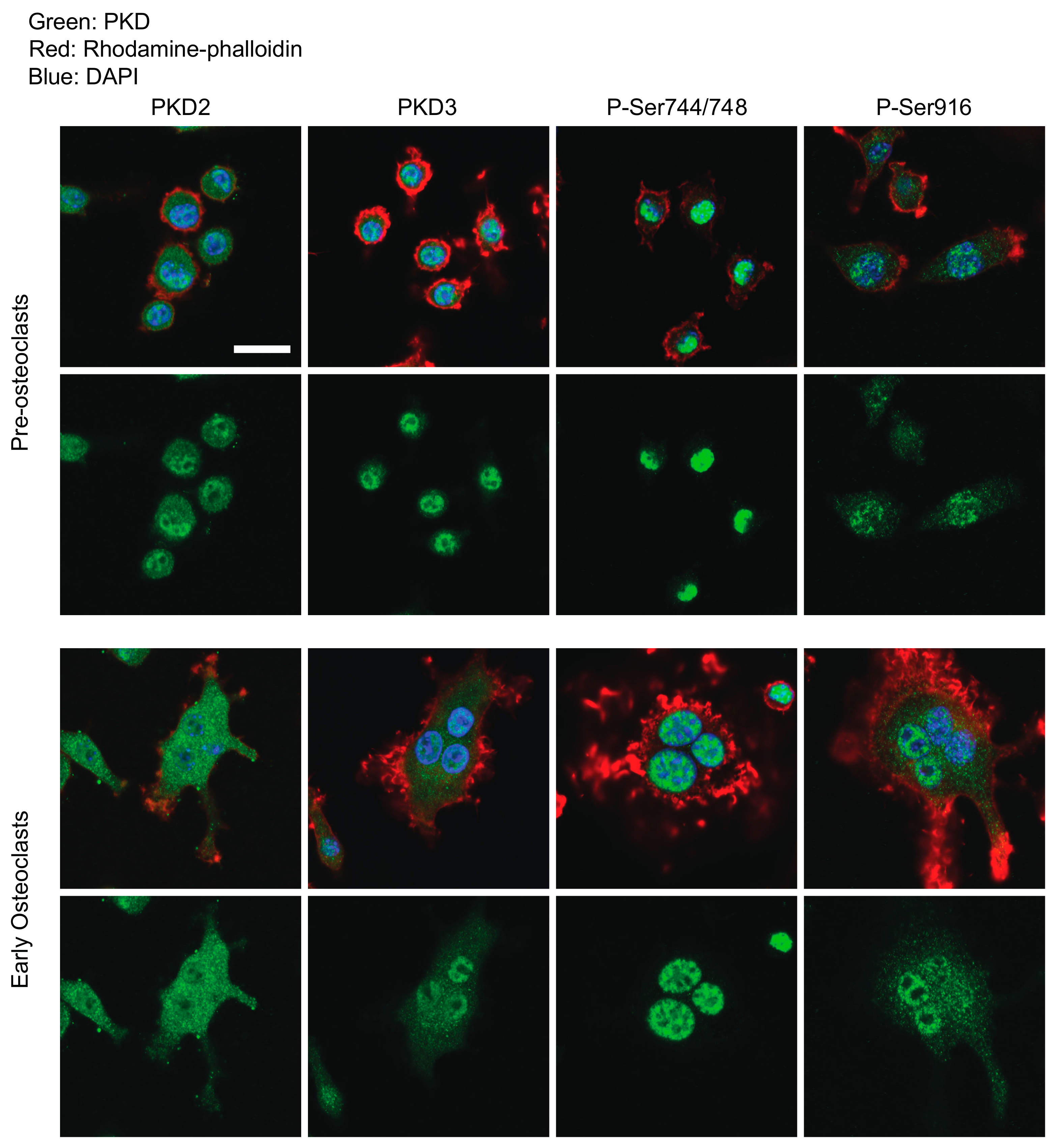
2.1. Determination of PKDs’ Subcellular Localization and Phosphorylation Through Osteoclastogenesis
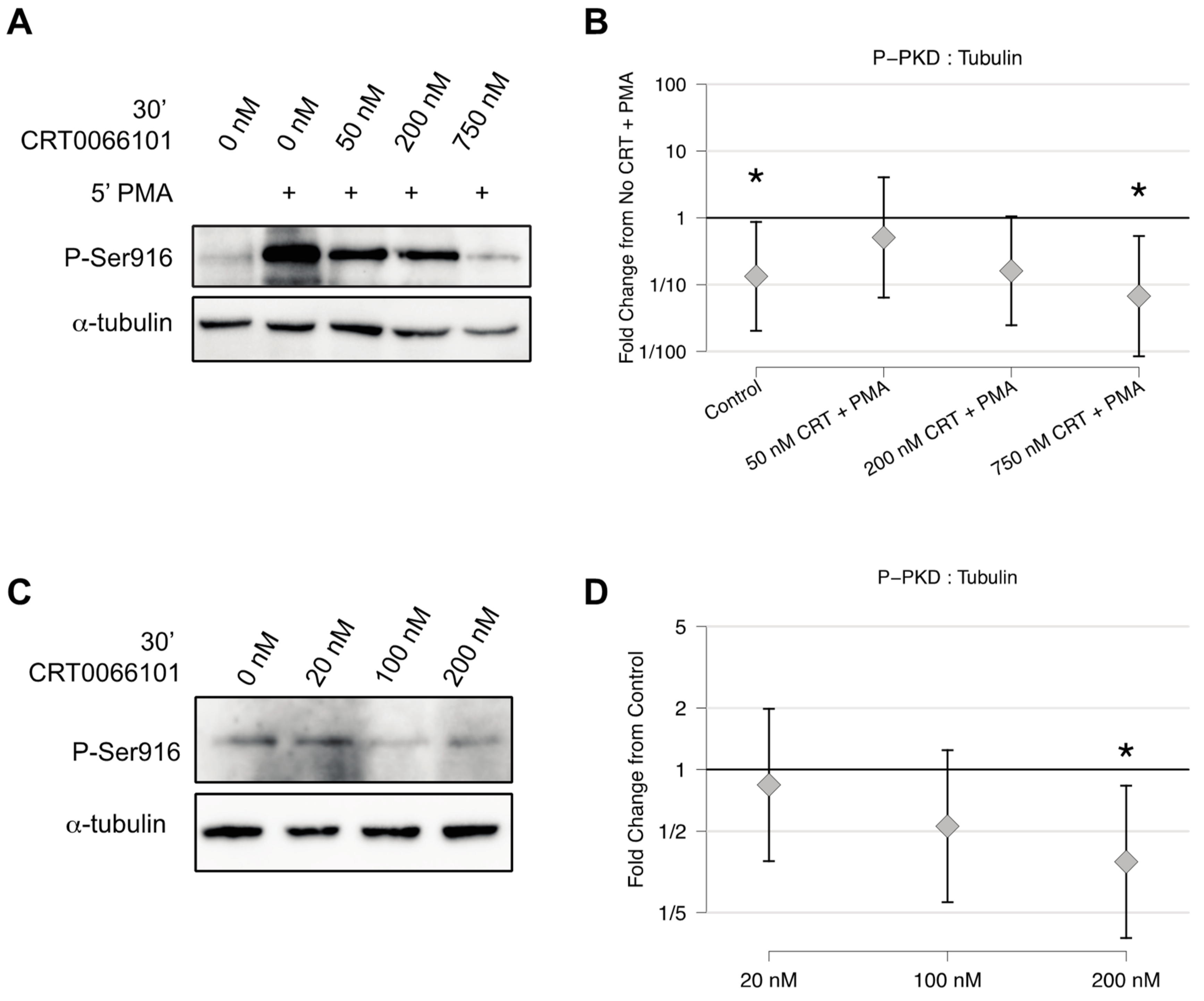
2.2. CRT0066101 Inhibits PKD Activity in Osteoclasts
2.3. CRT0066101 Inhibits in Vitro Osteoclast Differentiation
2.4. CRT0066101 Reduces Preosteoclast Motility
2.5. CRT0066101 Inhibits Osteoclast Resorptive Activity
2.6. Actin Belt Morphology in Mature Osteoclasts Requires PKD Function
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Mice and in Vitro Osteoclast Culture
4.3. Immunofluorescence Microscopy
4.4. Western Blotting
4.5. TRAP Staining
4.6. Caspase 3/7 Assay
4.7. Toxicity Assay
4.8. Quantitative RT-PCR
4.9. Transwell Migration Assay
4.10. Scratch Wound Migration Assay
4.11. Resorption Assays
4.12. Experimental Replication and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RANKL | Receptor Activator of NF-kB Ligand |
| M-CSF | Macrophage Colony Stimulating Factor |
| TRAP | Tartrate-Resistant Acid Phosphatase |
| PKD | Protein Kinase D |
| BMM | Bone Marrow Macrophage |
| PMA | Phorbol 12-myristate 13-acetate |
References
- Boyce, B.F. Advances in the regulation of osteoclasts and osteoclast functions. J. Dent. Res. 2013, 92, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashid, M.; Ramkumar, D.B.; Raskin, K.; Schwab, J.; Hornicek, F.J.; Lozano-Calderon, S.A. Paget Disease of Bone. Orthop. Clin. N. Am. 2015, 46, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Vallet, M.; Ralston, S.H. Biology and Treatment of Paget’s Disease of Bone. J. Cell. Biochem. 2016, 117, 289–299. [Google Scholar] [CrossRef]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of Bone Resorption in Periodontitis. J. Immunol. Res. 2015, 2015, 615486. [Google Scholar] [CrossRef]
- Schett, G.; Gravallese, E. Bone erosion in rheumatoid arthritis: Mechanisms, diagnosis and treatment. Nat. Rev. Rheumatol. 2012, 8, 656–664. [Google Scholar] [CrossRef]
- Shane, E.; Burr, D.; Abrahamsen, B.; Adler, R.A.; Brown, T.D.; Cheung, A.M.; Cosman, F.; Curtis, J.R.; Dell, R.; Dempster, D.W.; et al. Atypical subtrochanteric and diaphyseal femoral fractures: Second report of a task force of the American Society for Bone and Mineral Research. J. Bone Miner. Res. 2014, 29, 1–23. [Google Scholar] [CrossRef]
- Brown, J.P. Antiresorptives: Safety Concerns-Clinical Perspective. Toxicol. Pathol. 2017, 45, 859–863. [Google Scholar] [CrossRef]
- Gartrell, B.A.; Coleman, R.E.; Fizazi, K.; Miller, K.; Saad, F.; Sternberg, C.N.; Galsky, M.D. Toxicities following treatment with bisphosphonates and receptor activator of nuclear factor-kappaB ligand inhibitors in patients with advanced prostate cancer. Eur. Urol. 2014, 65, 278–286. [Google Scholar] [CrossRef]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef]
- Pereira, M.; Petretto, E.; Gordon, S.; Bassett, J.H.D.; Williams, G.R.; Behmoaras, J. Common signalling pathways in macrophage and osteoclast multinucleation. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Takito, J.; Inoue, S.; Nakamura, M. The Sealing Zone in Osteoclasts: A Self-Organized Structure on the Bone. Int. J. Mol. Sci. 2018, 19, 984. [Google Scholar] [CrossRef] [PubMed]
- Luxenburg, C.; Geblinger, D.; Klein, E.; Anderson, K.; Hanein, D.; Geiger, B.; Addadi, L. The architecture of the adhesive apparatus of cultured osteoclasts: From podosome formation to sealing zone assembly. PLoS ONE 2007, 2, e179. [Google Scholar] [CrossRef] [PubMed]
- Georgess, D.; Machuca-Gayet, I.; Blangy, A.; Jurdic, P. Podosome organization drives osteoclast-mediated bone resorption. Cell Adhes. Migr. 2014, 8, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Destaing, O.; Saltel, F.; Geminard, J.C.; Jurdic, P.; Bard, F. Podosomes display actin turnover and dynamic self-organization in osteoclasts expressing actin-green fluorescent protein. Mol. Biol. Cell 2003, 14, 407–416. [Google Scholar] [CrossRef]
- Rykx, A.; De Kimpe, L.; Mikhalap, S.; Vantus, T.; Seufferlein, T.; Vandenheede, J.R.; Van Lint, J. Protein kinase D: A family affair. FEBS Lett. 2003, 546, 81–86. [Google Scholar] [CrossRef]
- Rozengurt, E. Protein kinase D signaling: Multiple biological functions in health and disease. Physiology (Bethesda) 2011, 26, 23–33. [Google Scholar] [CrossRef]
- Waldron, R.T.; Rozengurt, E. Protein kinase C phosphorylates protein kinase D activation loop Ser744 and Ser748 and releases autoinhibition by the pleckstrin homology domain. J. Biol. Chem. 2003, 278, 154–163. [Google Scholar] [CrossRef]
- Jacamo, R.; Sinnett-Smith, J.; Rey, O.; Waldron, R.T.; Rozengurt, E. Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: Differential regulation of activation loop Ser(744) and Ser(748) phosphorylation. J. Biol. Chem. 2008, 283, 12877–12887. [Google Scholar] [CrossRef]
- Sinnett-Smith, J.; Jacamo, R.; Kui, R.; Wang, Y.M.; Young, S.H.; Rey, O.; Waldron, R.T.; Rozengurt, E. Protein kinase D mediates mitogenic signaling by Gq-coupled receptors through protein kinase C-independent regulation of activation loop Ser744 and Ser748 phosphorylation. J. Biol. Chem. 2009, 284, 13434–13445. [Google Scholar] [CrossRef]
- Waldron, R.T.; Innamorati, G.; Torres-Marquez, M.E.; Sinnett-Smith, J.; Rozengurt, E. Differential PKC-dependent and -independent PKD activation by G protein alpha subunits of the Gq family: Selective stimulation of PKD Ser(7)(4)(8) autophosphorylation by Galphaq. Cell Signal. 2012, 24, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Rubin, C.S. Protein kinase D: Coupling extracellular stimuli to the regulation of cell physiology. EMBO Rep. 2011, 12, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Ye, J.; Deng, F.; Wang, Q.J. Protein kinase D signaling in cancer: A friend or foe? Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Mansky, K.C.; Jensen, E.D.; Davidova, J.; Yamamoto, M.; Gopalakrishnan, R. Protein kinase D promotes in vitro osteoclast differentiation and fusion. J. Biol. Chem. 2013, 288, 9826–9834. [Google Scholar] [CrossRef]
- Papazyan, R.; Doche, M.; Waldron, R.T.; Rozengurt, E.; Moyer, M.P.; Rey, O. Protein kinase D isozymes activation and localization during mitosis. Exp. Cell Res. 2008, 314, 3057–3068. [Google Scholar] [CrossRef][Green Version]
- Fukunaga, T.; Zou, W.; Warren, J.T.; Teitelbaum, S.L. Vinculin regulates osteoclast function. J. Biol. Chem. 2014, 289, 13554–13564. [Google Scholar] [CrossRef]
- Harikumar, K.B.; Kunnumakkara, A.B.; Ochi, N.; Tong, Z.; Deorukhkar, A.; Sung, B.; Kelland, L.; Jamieson, S.; Sutherland, R.; Raynham, T.; et al. A novel small-molecule inhibitor of protein kinase D blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1136–1146. [Google Scholar] [CrossRef]
- Borges, S.; Perez, E.A.; Thompson, E.A.; Radisky, D.C.; Geiger, X.J.; Storz, P. Effective Targeting of Estrogen Receptor-Negative Breast Cancers with the Protein Kinase D Inhibitor CRT0066101. Mol. Cancer. Ther. 2015, 14, 1306–1316. [Google Scholar] [CrossRef]
- Hao, F.; Xu, Q.; Zhao, Y.; Stevens, J.V.; Young, S.H.; Sinnett-Smith, J.; Rozengurt, E. Insulin Receptor and GPCR Crosstalk Stimulates YAP via PI3K and PKD in Pancreatic Cancer Cells. Mol. Cancer Res. 2017, 15, 929–941. [Google Scholar] [CrossRef]
- Jensen, D.D.; Zhao, P.; Jimenez-Vargas, N.N.; Lieu, T.; Gerges, M.; Yeatman, H.R.; Canals, M.; Vanner, S.J.; Poole, D.P.; Bunnett, N.W. Protein Kinase D and Gbetagamma Subunits Mediate Agonist-evoked Translocation of Protease-activated Receptor-2 from the Golgi Apparatus to the Plasma Membrane. J. Biol. Chem. 2016, 291, 11285–11299. [Google Scholar] [CrossRef]
- Ni, Y.; Sinnett-Smith, J.; Young, S.H.; Rozengurt, E. PKD1 mediates negative feedback of PI3K/Akt activation in response to G protein-coupled receptors. PLoS ONE 2013, 8, e73149. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A.; Barisic, S.; Hausser, A. Multi-level control of actin dynamics by protein kinase D. Cell Signal. 2013, 25, 1739–1747. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Saini, S.; Liu, T.; Yoo, J. Bradykinin stimulates protein kinase D-mediated colonic myofibroblast migration via cyclooxygenase-2 and heat shock protein 27. J. Surg. Res. 2017, 209, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Torres-Marquez, E.; Sinnett-Smith, J.; Guha, S.; Kui, R.; Waldron, R.T.; Rey, O.; Rozengurt, E. CID755673 enhances mitogenic signaling by phorbol esters, bombesin and EGF through a protein kinase D-independent pathway. Biochem. Biophys. Res. Commun. 2010, 391, 63–68. [Google Scholar] [CrossRef]
- Yuan, J.; Tan, T.; Geng, M.; Tan, G.; Chheda, C.; Pandol, S.J. Novel Small Molecule Inhibitors of Protein Kinase D Suppress NF-kappaB Activation and Attenuate the Severity of Rat Cerulein Pancreatitis. Front. Physiol. 2017, 8, 1014. [Google Scholar] [CrossRef]
- Bowden, E.T.; Barth, M.; Thomas, D.; Glazer, R.I.; Mueller, S.C. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene 1999, 18, 4440–4449. [Google Scholar] [CrossRef]
- Bastea, L.I.; Doppler, H.; Balogun, B.; Storz, P. Protein kinase D1 maintains the epithelial phenotype by inducing a DNA-bound, inactive SNAI1 transcriptional repressor complex. PLoS ONE 2012, 7, e30459. [Google Scholar] [CrossRef]
- Du, C.; Zhang, C.; Hassan, S.; Biswas, M.H.; Balaji, K.C. Protein kinase D1 suppresses epithelial-to-mesenchymal transition through phosphorylation of snail. Cancer Res. 2010, 70, 7810–7819. [Google Scholar] [CrossRef]
- Eiseler, T.; Kohler, C.; Nimmagadda, S.C.; Jamali, A.; Funk, N.; Joodi, G.; Storz, P.; Seufferlein, T. Protein kinase D1 mediates anchorage-dependent and -independent growth of tumor cells via the zinc finger transcription factor Snail1. J. Biol. Chem. 2012, 287, 32367–32380. [Google Scholar] [CrossRef]
- Bernhart, E.; Damm, S.; Wintersperger, A.; DeVaney, T.; Zimmer, A.; Raynham, T.; Ireson, C.; Sattler, W. Protein kinase D2 regulates migration and invasion of U87MG glioblastoma cells in vitro. Exp. Cell Res. 2013, 319, 2037–2048. [Google Scholar] [CrossRef]
- Sinnett-Smith, J.; Zhukova, E.; Rey, O.; Rozengurt, E. Protein kinase D2 potentiates MEK/ERK/RSK signaling, c-Fos accumulation and DNA synthesis induced by bombesin in Swiss 3T3 cells. J. Cell. Physiol. 2007, 211, 781–790. [Google Scholar] [CrossRef]
- Yamashita, K.; Gon, Y.; Shimokawa, T.; Nunomura, S.; Endo, D.; Miyata, N.; Hashimoto, S.; Van Lint, J.; Ra, C. High affinity receptor for IgE stimulation activates protein kinase D augmenting activator protein-1 activity for cytokine producing in mast cells. Int. Immunopharmacol. 2010, 10, 277–283. [Google Scholar] [CrossRef]
- Zhang, W.; Zheng, S.; Storz, P.; Min, W. Protein kinase D specifically mediates apoptosis signal-regulating kinase 1-JNK signaling induced by H2O2 but not tumor necrosis factor. J. Biol. Chem. 2005, 280, 19036–19044. [Google Scholar] [CrossRef]
- Parra, M.; Kasler, H.; McKinsey, T.A.; Olson, E.N.; Verdin, E. Protein kinase D1 phosphorylates HDAC7 and induces its nuclear export after T-cell receptor activation. J. Biol. Chem. 2005, 280, 13762–13770. [Google Scholar] [CrossRef]
- Vega, R.B.; Harrison, B.C.; Meadows, E.; Roberts, C.R.; Papst, P.J.; Olson, E.N.; McKinsey, T.A. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell Biol. 2004, 24, 8374–8385. [Google Scholar] [CrossRef]
- Brandlin, I.; Hubner, S.; Eiseler, T.; Martinez-Moya, M.; Horschinek, A.; Hausser, A.; Link, G.; Rupp, S.; Storz, P.; Pfizenmaier, K.; et al. Protein kinase C (PKC)eta-mediated PKC mu activation modulates ERK and JNK signal pathways. J. Biol. Chem. 2002, 277, 6490–6496. [Google Scholar] [CrossRef]
- Hausser, A.; Storz, P.; Hubner, S.; Braendlin, I.; Martinez-Moya, M.; Link, G.; Johannes, F.J. Protein kinase C mu selectively activates the mitogen-activated protein kinase (MAPK) p42 pathway. FEBS Lett. 2001, 492, 39–44. [Google Scholar] [CrossRef]
- Lemonnier, J.; Ghayor, C.; Guicheux, J.; Caverzasio, J. Protein kinase C-independent activation of protein kinase D is involved in BMP-2-induced activation of stress mitogen-activated protein kinases JNK and p38 and osteoblastic cell differentiation. J. Biol. Chem. 2004, 279, 259–264. [Google Scholar] [CrossRef]
- Eisenberg-Lerner, A.; Kimchi, A. DAP kinase regulates JNK signaling by binding and activating protein kinase D under oxidative stress. Cell Death Differ. 2007, 14, 1908–1915. [Google Scholar] [CrossRef]
- Yamamoto, H.; Oue, N.; Sato, A.; Hasegawa, Y.; Yamamoto, H.; Matsubara, A.; Yasui, W.; Kikuchi, A. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene 2010, 29, 2036–2046. [Google Scholar] [CrossRef]
- Storz, P.; Doppler, H.; Toker, A. Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol. Cell Biol. 2004, 24, 2614–2626. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Toker, A. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J. 2003, 22, 109–120. [Google Scholar] [CrossRef]
- Roy, A.; Veroli, M.V.; Prasad, S.; Wang, Q.J. Protein Kinase D2 Modulates Cell Cycle By Stabilizing Aurora A Kinase at Centrosomes. Mol. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Biswas, M.H.; Zhang, C.; Du, C.; Balaji, K.C. Heat shock protein 27 mediates repression of androgen receptor function by protein kinase D1 in prostate cancer cells. Oncogene 2009, 28, 4386–4396. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Gao, Y.; Zhang, L.; Weng, Z.; Zhang, F.; Hu, X.; Wang, S.; Vosler, P.; Cao, G.; Sun, D.; et al. Phosphorylation of HSP27 by protein kinase D is essential for mediating neuroprotection against ischemic neuronal injury. J. Neurosci. 2012, 32, 2667–2682. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Rozengurt, E. PKD, PKD2, and p38 MAPK mediate Hsp27 serine-82 phosphorylation induced by neurotensin in pancreatic cancer PANC-1 cells. J. Cell. Biochem. 2008, 103, 648–662. [Google Scholar] [CrossRef]
- Ford, J.J.; Yeh, L.C.; Schmidgal, E.C.; Thompson, J.F.; Adamo, M.L.; Lee, J.C. Protein kinase D1 is essential for bone acquisition during pubertal growth. Endocrinology 2013, 154, 4182–4191. [Google Scholar] [CrossRef][Green Version]
- Li, S.; Xu, W.; Xing, Z.; Qian, J.; Chen, L.; Gu, R.; Guo, W.; Lai, X.; Zhao, W.; Wang, Y.; et al. A Conditional Knockout Mouse Model Reveals a Critical Role of PKD1 in Osteoblast Differentiation and Bone Development. Sci. Rep. 2017, 7, 40505. [Google Scholar] [CrossRef]
- Ellwanger, K.; Hausser, A. Physiological functions of protein kinase D in vivo. IUBMB Life 2013, 65, 98–107. [Google Scholar] [CrossRef]
- Xing, L.; Boyce, B.F. RANKL-based osteoclastogenic assays from murine bone marrow cells. Methods Mol. Biol. 2014, 1130, 307–313. [Google Scholar] [CrossRef]
- Takeshita, S.; Kaji, K.; Kudo, A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J. Bone Miner. Res. 2000, 15, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Orriss, I.R.; Arnett, T.R. Rodent osteoclast cultures. Methods Mol. Biol. 2012, 816, 103–117. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leightner, A.C.; Mello Guimaraes Meyers, C.; Evans, M.D.; Mansky, K.C.; Gopalakrishnan, R.; Jensen, E.D. Regulation of Osteoclast Differentiation at Multiple Stages by Protein Kinase D Family Kinases. Int. J. Mol. Sci. 2020, 21, 1056. https://doi.org/10.3390/ijms21031056
Leightner AC, Mello Guimaraes Meyers C, Evans MD, Mansky KC, Gopalakrishnan R, Jensen ED. Regulation of Osteoclast Differentiation at Multiple Stages by Protein Kinase D Family Kinases. International Journal of Molecular Sciences. 2020; 21(3):1056. https://doi.org/10.3390/ijms21031056
Chicago/Turabian StyleLeightner, Amanda C., Carina Mello Guimaraes Meyers, Michael D. Evans, Kim C. Mansky, Rajaram Gopalakrishnan, and Eric D. Jensen. 2020. "Regulation of Osteoclast Differentiation at Multiple Stages by Protein Kinase D Family Kinases" International Journal of Molecular Sciences 21, no. 3: 1056. https://doi.org/10.3390/ijms21031056
APA StyleLeightner, A. C., Mello Guimaraes Meyers, C., Evans, M. D., Mansky, K. C., Gopalakrishnan, R., & Jensen, E. D. (2020). Regulation of Osteoclast Differentiation at Multiple Stages by Protein Kinase D Family Kinases. International Journal of Molecular Sciences, 21(3), 1056. https://doi.org/10.3390/ijms21031056