CD155: A Multi-Functional Molecule in Tumor Progression

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
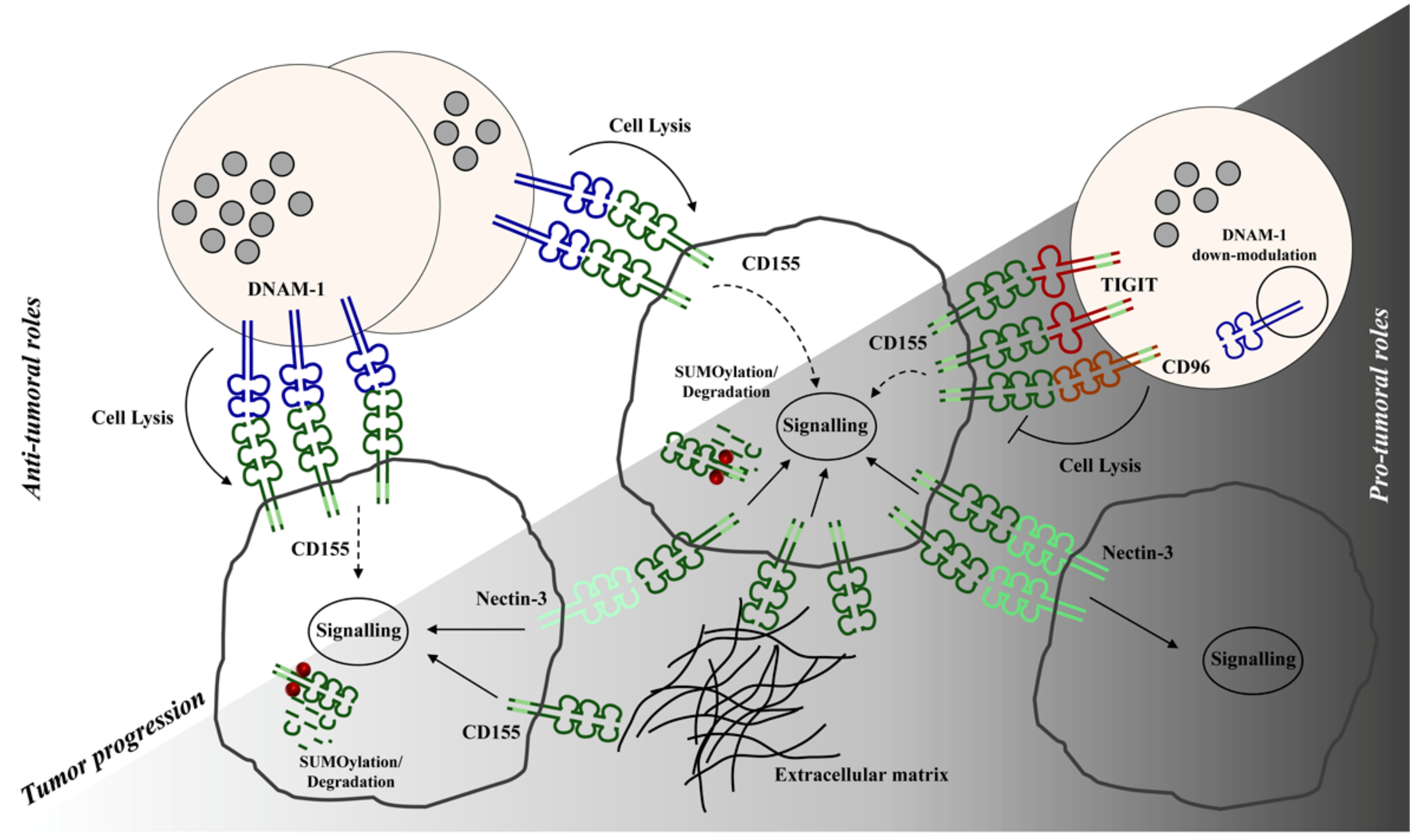
2. CD155-Mediated Signals Promote Tumor Progression
3. CD155 is a Ligand for Immunoreceptors Implicated in Tumor Surveillance
3.1. Interaction of CD155 with DNAM-1 Activating Receptor
3.2. Inhibitory CD155 Receptors: TIGIT and CD96
4. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Fuchs, A.; Colonna, M. The role of NK cell recognition of nectin and nectin-like proteins in tumor immunosurveillance. Seminm. Cancer Biol. 2006, 16, 359–366. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Madore, J.; Li, X.Y.; Smyth, M.J. Tumor intrinsic and extrinsic immune functions of CD155. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Bowers, J.R.; Readler, J.M.; Sharma, P.; Excoffon, K.J.D.A. Poliovirus Receptor: More than a simple viral receptor. Virus Res. 2017, 242, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, C.L.; Wimmer, E.; Racaniello, V.R. Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 1989, 56, 855–865. [Google Scholar] [CrossRef]
- Sakisaka, T.; Ikeda, W.; Ogita, H.; Fujita, N.; Takai, Y. The roles of nectins in cell adhesions: Cooperation with other cell adhesion molecules and growth factor receptors. Curr. Opin. Cell Biol. 2007, 19, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Rikitake, Y.; Mandai, K.; Takai, Y. The role of nectins in different types of cell-cell adhesion. J. Cell Sci. 2012, 125, 3713–3722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, Y.; Ikeda, W.; Ogita, H.; Rikitake, Y. The immunoglobulin-like cell adhesion molecule nectin and its associated protein afadin. Annu. Rev. Cell Dev. Biol. 2008, 24, 309–342. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Cao, X.; Welker, R.; Wimmer, E. Interaction of the poliovirus receptor CD155 with the dynein light chain Tctex-1 and its implication for poliovirus pathogenesis. J. Biol. Chem. 2002, 277, 7897–7904. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, W.; Kakunaga, S.; Itoh, S.; Shingai, T.; Takekuni, K.; Satoh, K.; Inoue, Y.; Hamaguchi, A.; Morimoto, K.; Takeuchi, M.; et al. Tage4/Nectin-like molecule-5 heterophilically trans-interacts with cell adhesion molecule Nectin-3 and enhances cell migration. J. Biol. Chem. 2003, 278, 28167–28172. [Google Scholar] [CrossRef] [Green Version]
- Mueller, S.; Wimmer, E. Recruitment of nectin-3 to cell-cell junctions through trans-heterophilic interaction with CD155, a vitronectin and poliovirus receptor that localizes to alpha(v)beta3 integrin-containing membrane microdomains. J. Biol. Chem. 2003, 278, 31251–31260. [Google Scholar] [CrossRef] [Green Version]
- Lange, R.; Peng, X.; Wimmer, E.; Lipp, M.; Bernhardt, G. The poliovirus receptor CD155 mediates cell-to-matrix contacts by specifically binding to vitronectin. Virology 2001, 285, 218–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, S.; Horie, H.; Ise, I.; Okitsu, A.; Yoshida, M.; Iizuka, N.; Takeuchi, K.; Takegami, T.; Nomoto, A. The poliovirus receptor protein is produced both as membrane-bound and secreted forms. EMBO J. 1990, 9, 3217–3224. [Google Scholar] [CrossRef] [PubMed]
- Baury, B.; Masson, D.; McDermott, B.M., Jr.; Jarry, A.; Blottière, H.M.; Blanchardie, P.; Laboisse, C.L.; Lustenberger, P.; Racaniello, V.R.; Denis, M.G. Identification of secreted CD155 isoforms. Biochem. Biophys. Res. Commun. 2003, 309, 175–182. [Google Scholar] [CrossRef]
- Ohka, S.; Ohno, H.; Tohyama, K.; Nomoto, A. Basolateral sorting of human poliovirus receptor alpha involves an interaction with the mu1B subunit of the clathrin adaptor complex in polarized epithelial cells. Biochem. Biophys. Res. Commun. 2001, 287, 941–948. [Google Scholar] [CrossRef]
- Oda, T.; Ohka, S.; Nomoto, A. Ligand stimulation of CD155alpha inhibits cell adhesion and enhances cell migration in fibroblasts. Biochem. Biophys. Res. Commun. 2004, 319, 1253–1264. [Google Scholar] [CrossRef]
- Masson, D.; Jarry, A.; Baury, B.; Blanchardie, P.; Laboisse, C.; Lustenberger, P.; Denis, M.G. Overexpression of the CD155 gene in human colorectal carcinoma. Gut 2001, 49, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Sloan, K.E.; Eustace, B.K.; Stewart, J.K.; Zehetmeier, C.; Torella, C.; Simeone, M.; Roy, J.E.; Unger, C.; Louis, D.N.; Ilag, L.L.; et al. CD155/CD155 plays a key role in cell motility during tumor cell invasion and migration. BMC Cancer 2004, 4, 73. [Google Scholar] [CrossRef] [Green Version]
- Nakai, R.; Maniwa, Y.; Tanaka, Y.; Nishio, W.; Yoshimura, M.; Okita, Y.; Ohbayashi, C.; Satoh, N.; Ogita, H.; Takai, Y.; et al. Overexpression of Necl-5 correlates with unfavorable prognosis in patients with lung adenocarcinoma. Cancer Sci. 2010, 101, 1326–1330. [Google Scholar] [CrossRef]
- Bevelacqua, V.; Bevelacqua, Y.; Candido, S.; Skarmoutsou, E.; Amoroso, A.; Guarneri, C.; Strazzanti, A.; Gangemi, P.; Mazzarino, M.C.; D’Amico, F.; et al. Nectin like-5 overexpression correlates with the malignant phenotype in cutaneous melanoma. Oncotarget 2012, 3, 882–892. [Google Scholar] [CrossRef] [Green Version]
- Nishiwada, S.; Sho, M.; Yasuda, S.; Shimada, K.; Yamato, I.; Akahori, T.; Kinoshita, S.; Nagai, M.; Konishi, N.; Nakajima, Y. Clinical significance of CD155 expression in human pancreatic cancer. Anticancer Res. 2015, 35, 2287–2297. [Google Scholar]
- Iguchi-Manaka, A.; Okumura, G.; Kojima, H.; Cho, Y.; Hirochika, R.; Bando, H.; Sato, T.; Yoshikawa, H.; Hara, H.; Shibuya, A.; et al. Increased soluble CD155 in the serum of cancer patients. PLoS ONE 2016, 11, e0152982. [Google Scholar] [CrossRef] [Green Version]
- de Andrade, L.F.; Smyth, M.J.; Martinet, L. DNAM-1 control of natural killer cells functions through nectin and nectin-like proteins. Immunol. Cell Biol. 2014, 92, 237–244. [Google Scholar] [CrossRef]
- Martinet, L.; Smyth, M.J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 2015, 15, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Hirota, T.; Irie, K.; Okamoto, R.; Ikeda, W.; Takai, Y. Transcriptional activation of the mouse Necl-5/Tage4/CD155/CD155 gene by fibroblast growth factor or oncogenic Ras through the Raf-MEK-ERK-AP-1 pathway. Oncogene 2005, 24, 2229–2235. [Google Scholar] [CrossRef] [Green Version]
- Solecki, D.J.; Gromeier, M.; Mueller, S.; Bernhardt, G.; Wimmer, E. Expression of the human poliovirus receptor/CD155 gene is activated by sonic hedgehog. J. Biol. Chem. 2002, 277, 25697–25702. [Google Scholar] [CrossRef] [Green Version]
- Kono, T.; Imai, Y.; Yasuda, S.; Ohmori, K.; Fukui, H.; Ichikawa, K.; Tomita, S.; Imura, J.; Kuroda, Y.; Ueda, Y.; et al. The CD155/poliovirus receptor enhances the proliferation of ras-mutated cells. Int. J. Cancer. 2008, 122, 317–324. [Google Scholar] [CrossRef]
- Kakunaga, S.; Ikeda, W.; Shingai, T.; Fujito, T.; Yamada, A.; Minami, Y.; Imai, T.; Takai, Y. Enhancement of serum- and platelet-derived growth factor-induced cell proliferation by Necl-5/Tage4/poliovirus receptor/CD155 through the Ras-Raf-MEK-ERK signaling. J. Biol. Chem. 2004, 279, 36419–36425. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Wang, B.; Gao, J.; Xin, N.; Wang, W.; Song, X.; Shao, Y.; Zhao, C. CD155 knockdown promotes apoptosis via AKT/Bcl-2/Bax in colon cancer cells. J. Cell. Mol. Med. 2018, 22, 131–140. [Google Scholar] [CrossRef]
- Sullivan, D.P.; Seidman, M.A.; Muller, W.A. Poliovirus receptor (CD155) regulates a step in transendothelial migration between PECAM and CD99. Am. J. Pathol. 2013, 182, 1031–1042. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Irie, K.; Okamoto, R.; Ooshio, T.; Fujita, N.; Takai, Y. Common signaling pathway is used by the trans-interaction of Necl-5/Tage4/CD155/CD155 and nectin, and of nectin and nectin during the formation of cell-cell adhesion. Cancer Sci. 2005, 96, 578–589. [Google Scholar] [CrossRef]
- Sloan, K.E.; Stewart, J.K.; Treloar, A.F.; Matthews, R.T.; Jay, D.G. CD155/CD155 enhances glioma cell dispersal by regulating adhesion signaling and focal adhesion dynamics. Cancer Res. 2005, 65, 10930–10937. [Google Scholar] [CrossRef] [Green Version]
- Fujito, T.; Ikeda, W.; Kakunaga, S.; Minami, Y.; Kajita, M.; Sakamoto, Y.; Monden, M.; Takai, Y. Inhibition of cell movement and proliferation by cell-cell contact-induced interaction of Necl-5 with nectin-3. J. Cell Biol. 2005, 171, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Abe, A.; Fukui, H.; Fujii, S.; Kono, T.; Mukawa, K.; Yoshitake, N.; Sekikawa, A.; Ichikawa, K.; Tomita, S.; Yamagishi, H.; et al. Role of Necl-5 in the pathophysiology of colorectal lesions induced by dimethylhydrazine and/or dextran sodium sulphate. J. Pathol. 2009, 217, 42–53. [Google Scholar] [CrossRef]
- Li, X.Y.; Das, I.; Lepletier, A.; Addala, V.; Bald, T.; Stannard, K.; Barkauskas, D.; Liu, J.; Aguilera, A.R.; Takeda, K.; et al. CD155 loss enhances tumor suppression via combined host and tumor-intrinsic mechanisms. J. Clin. Invest. 2018, 128, 2613–2625. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.C.; Dobrikova, E.Y.; Dobrikov, M.I.; Walton, R.W.; Gemberling, S.L.; Nair, S.K.; Desjardins, A.; Sampson, J.H.; Friedman, H.S.; Friedman, A.H.; et al. Oncolytic polio virotherapy of cancer. Cancer 2014, 120, 3277–3286. [Google Scholar] [CrossRef] [Green Version]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Chan, C.J.; Andrews, D.M.; Smyth, M.J. Receptors that interact with nectin and nectin-like proteins in the immunosurveillance and immunotherapy of cancer. Curr. Opin. Immunol. 2012, 24, 246–251. [Google Scholar] [CrossRef]
- Shibuya, K.; Lanier, L.L.; Phillips, J.H.; Ochs, H.D.; Shimizu, K.; Nakayama, E.; Nakauchi, H.; Shibuya, A. Physical and functional association of LFA-1 with DNAM-1 adhesion molecule. Immunity 1999, 11, 615–623. [Google Scholar] [CrossRef] [Green Version]
- Bryceson, Y.T.; March, M.E.; Ljunggren, H.G.; Long, E.O. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood 2006, 107, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Gilfillan, S.; Chan, C.J.; Cella, M.; Haynes, N.M.; Rapaport, A.S.; Boles, K.S.; Andrews, D.M.; Smyth, M.J.; Colonna, M. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J. Exp. Med. 2008, 205, 2965–2973. [Google Scholar] [CrossRef] [Green Version]
- Iguchi-Manaka, A.; Kai, H.; Yamashita, Y.; Shibata, K.; Tahara-Hanaoka, S.; Honda, S.; Yasui, T.; Kikutani, H.; Shibuya, K.; Shibuya, A. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J. Exp. Med. 2008, 205, 2959–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara-Hanaoka, S.; Shibuya, K.; Kai, H.; Miyamoto, A.; Morikawa, Y.; Ohkochi, N.; Honda, S.; Shibuya, A. Tumor rejection by the poliovirus receptor family ligands of the DNAM-1 (CD226) receptor. Blood 2006, 107, 1491–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmikanth, T.; Burke, S.; Ali, T.H.; Kimpfler, S.; Ursini, F.; Ruggeri, L.; Capanni, M.; Umansky, V.; Paschen, A.; Sucker, A.; et al. NCRs and DNAM-1 mediate NK cell recognition and lysis of human and mouse melanoma cell lines in vitro and in vivo. J. Clin. Invest. 2009, 119, 1251–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.J.; Andrews, D.M.; McLaughlin, N.M.; Yagita, H.; Gilfillan, S.; Colonna, M.; Smyth, M.J. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J. Immunol. 2010, 184, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Croxford, J.L.; Tang, M.L.; Pan, M.F.; Huang, C.W.; Kamran, N.; Phua, C.M.; Chng, W.J.; Ng, S.B.; Raulet, D.H.; Gasser, S. ATM-dependent spontaneous regression of early Eμ-myc-induced murine B-cell leukemia depends on natural killer and T cells. Blood 2013, 121, 2512–2521. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.J.; Martinet, L.; Gilfillan, S.; Souza-Fonseca-Guimaraes, F.; Chow, M.T.; Town, L.; Ritchie, D.S.; Colonna, M.; Andrews, D.M.; Smyth, M.J. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat. Immunol. 2014, 15, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Guillerey, C.; Ferrari de Andrade, L.; Vuckovic, S.; Miles, K.; Ngiow, S.F.; Yong, M.C.; Teng, M.W.; Colonna, M.; Ritchie, D.S.; Chesi, M.; et al. Immunosurveillance and therapy of multiple myeloma are CD226 dependent. J. Clin. Invest. 2015, 125, 2077–2089. [Google Scholar] [CrossRef] [Green Version]
- Castriconi, R.; Dondero, A.; Corrias, M.V.; Lanino, E.; Pende, D.; Moretta, L.; Bottino, C.; Moretta, A. Natural killer cell-mediated killing of freshly isolated neuroblastoma cells: Critical role of DNAX accessory molecule-1-poliovirus receptor interaction. Cancer Res. 2004, 64, 9180–9184. [Google Scholar] [CrossRef] [Green Version]
- Pende, D.; Bottino, C.; Castriconi, R.; Cantoni, C.; Marcenaro, S.; Rivera, P.; Spaggiari, G.M.; Dondero, A.; Carnemolla, B.; Reymond, N.; et al. CD155 (CD155) and Nectin-2 (CD112) as ligands of the human DNAM-1 (CD226) activating receptor: Involvement in tumor cell lysis. Mol. Immunol. 2005, 42, 463–469. [Google Scholar] [CrossRef]
- Carlsten, M.; Björkström, N.K.; Norell, H.; Bryceson, Y.; van Hall, T.; Baumann, B.C.; Hanson, M.; Schedvins, K.; Kiessling, R.; Ljunggren, H.G.; et al. DNAX accessory molecule-1 mediated recognition of freshly isolated ovarian carcinoma by resting natural killer cells. Cancer Res. 2007, 67, 1317–1325. [Google Scholar] [CrossRef] [Green Version]
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007, 67, 8444–8449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriani, A.; Zingoni, A.; Cerboni, C.; Iannitto, M.L.; Ricciardi, M.R.; Di Gialleonardo, V.; Cippitelli, M.; Fionda, C.; Petrucci, M.T.; Guarini, A.; et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 2009, 113, 3503–3511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriani, A.; Iannitto, M.L.; Ricci, B.; Fionda, C.; Malgarini, G.; Morrone, S.; Peruzzi, G.; Ricciardi, M.R.; Petrucci, M.T.; Cippitelli, M.; et al. Reactive oxygen species- and DNA damage response-dependent NK cell activating ligand upregulation occurs at transcriptional levels and requires the transcriptional factor E2F1. J. Immunol. 2014, 193, 950–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerboni, C.; Fionda, C.; Soriani, A.; Zingoni, A.; Doria, M.; Cippitelli, M.; Santoni, A. The DNA Damage Response: A Common Pathway in the Regulation of NKG2D and DNAM-1 Ligand Expression in Normal, Infected, and Cancer Cells. Front. Immunol. 2014, 4, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fionda, C.; Abruzzese, M.P.; Zingoni, A.; Soriani, A.; Ricci, B.; Molfetta, R.; Paolini, R.; Santoni, A.; Cippitelli, M. Nitric oxide donors increase CD155/CD155 DNAM-1 ligand expression in multiple myeloma cells: Role of DNA damage response activation. BMC Cancer 2015, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Fionda, C.; Abruzzese, M.P.; Zingoni, A.; Cecere, F.; Vulpis, E.; Peruzzi, G.; Soriani, A.; Molfetta, R.; Paolini, R.; Ricciardi, M.R.; et al. The IMiDs targets IKZF-1/3 and IRF4 as novel negative regulators of NK cell-activating ligands expression in multiple myeloma. Oncotarget 2015, 6, 23609–23630. [Google Scholar] [CrossRef] [Green Version]
- Molfetta, R.; Zingoni, A.; Santoni, A.; Paolini, R. Post-translational Mechanisms Regulating NK Cell Activating Receptors and Their Ligands in Cancer: Potential Targets for Therapeutic Intervention. Front. Immunol. 2019, 10, 2557. [Google Scholar] [CrossRef] [Green Version]
- Balagopalan, L.; Barr, V.A.; Samelson, L.E. Endocytic events in TCR signaling: Focus on adapters in microclusters. Immunol. Rev. 2009, 232, 84–98. [Google Scholar] [CrossRef] [Green Version]
- Molfetta, R.; Gasparrini, F.; Santoni, A.; Paolini, R. Ubiquitination and endocytosis of the high affinity receptor for IgE. Mol. Immunol. 2010, 47, 2427–2434. [Google Scholar] [CrossRef]
- Drake, J.R. The immunobiology of ubiquitin-dependent B cell receptor functions. Mol. Immunol. 2018, 101, 146–154. [Google Scholar] [CrossRef]
- Gong, J.; Fang, L.; Liu, R.; Wang, Y.; Xing, J.; Chen, Y.; Zhuang, R.; Zhang, Y.; Zhang, C.; Yang, A.; et al. UPR decreases CD226 ligand CD155 expression and sensitivity to NK cell-mediated cytotoxicity in hepatoma cells. Eur. J. Immunol. 2014, 44, 3758–3767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
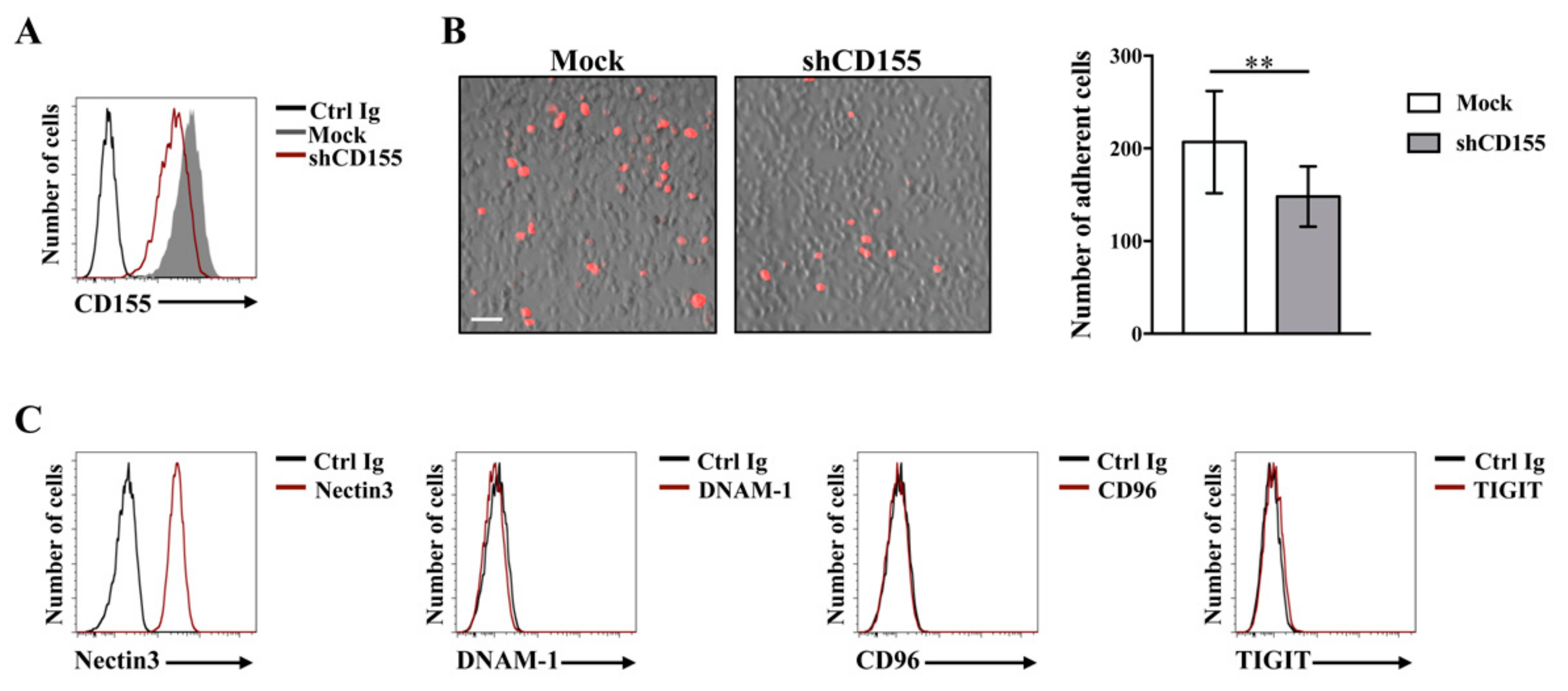
- Molfetta, R.; Milito, N.D.; Zitti, B.; Lecce, M.; Fionda, C.; Cippitelli, M.; Santoni, A.; Paolini, R. The Ubiquitin-proteasome pathway regulates Nectin2/CD112 expression and impairs NK cell recognition and killing. Eur. J. Immunol. 2019, 49, 873–883. [Google Scholar] [CrossRef] [PubMed]
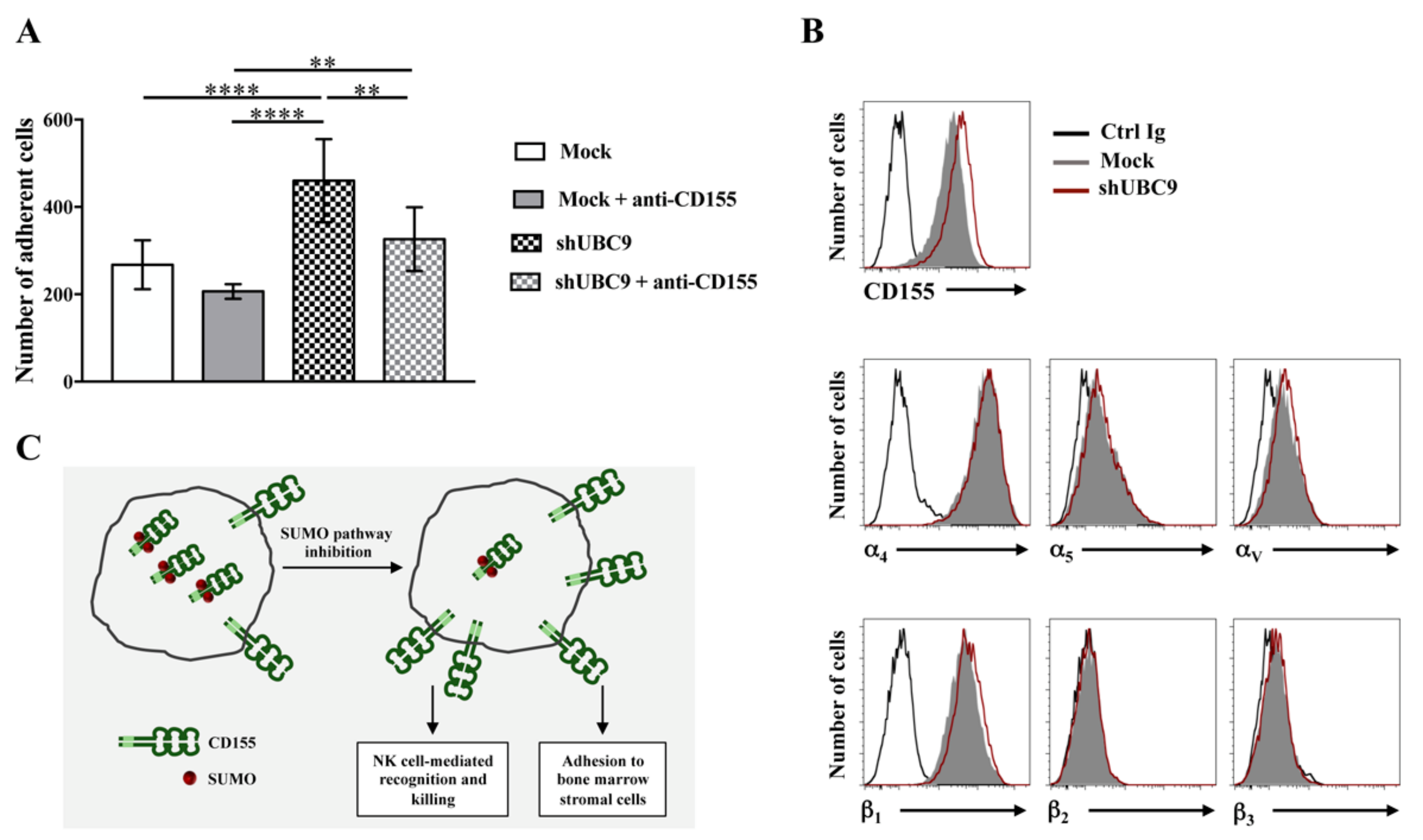
- Zitti, B.; Molfetta, R.; Fionda, C.; Quatrini, L.; Stabile, H.; Lecce, M.; de Turris, V.; Ricciardi, M.R.; Petrucci, M.T.; Cippitelli, M.; et al. Innate immune activating ligand SUMOylation affects tumor cell recognition by NK cells. Sci. Rep. 2017, 7, 10445. [Google Scholar] [CrossRef] [PubMed]
- Redondo-Muñoz, J.; García-Pardo, A.; Teixidó, J. Molecular Players in Hematologic Tumor Cell Trafficking. Front. Immunol. 2019, 10, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Correa, B.; Gayoso, I.; Bergua, J.M.; Casado, J.G.; Morgado, S.; Solana, R.; Tarazona, R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol. Cell Biol. 2012, 90, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Vulpis, E.; Stabile, H.; Soriani, A.; Fionda, C.; Petrucci, M.T.; Mariggio’, E.; Ricciardi, M.R.; Cippitelli, M.; Gismondi, A.; Santoni, A.; et al. Key Role of the CD56lowCD16low Natural Killer Cell Subset in the Recognition and Killing of Multiple Myeloma Cells. Cancers 2018, 10, 473. [Google Scholar] [CrossRef] [Green Version]
- Seth, S.; Qiu, Q.; Danisch, S.; Maier, M.K.; Braun, A.; Ravens, I.; Czeloth, N.; Hyde, R.; Dittrich-Breiholz, O.; Förster, R.; et al. Intranodal interaction with dendritic cells dynamically regulates surface expression of the co-stimulatory receptor CD226 protein on murine T cells. J. Biol. Chem. 2011, 286, 39153–39163. [Google Scholar] [CrossRef] [Green Version]
- Dougall, W.C.; Kurtulus, S.; Smyth, M.J.; Anderson, A.C. TIGIT and CD96: New checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017, 276, 112–120. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with CD155 and CD155L2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef]
- Stanietsky, N.; Rovis, T.L.; Glasner, A.; Seidel, E.; Tsukerman, P.; Yamin, R.; Enk, J.; Jonjic, S.; Mandelboim, O. Mouse TIGIT inhibits NK-cell cytotoxicity upon interaction with CD155. Eur. J. Immunol. 2013, 43, 2138–2150. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Stannard, K.; Liu, J.; Allen, S.; Yong, M.C.; Mittal, D.; Aguilera, A.R.; Miles, J.J.; Lutzky, V.P.; de Andrade, L.F.; et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov. 2016, 6, 446–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, A.; Cella, M.; Giurisato, E.; Shaw, A.S.; Colonna, M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155). J. Immunol. 2004, 172, 3994–3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgiev, H.; Ravens, I.; Papadogianni, G.; Bernhardt, G. Coming of Age: CD96 Emerges as Modulator of Immune Responses. Front. Immunol. 2018, 9, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, D.; Seth, S.; Albrecht, J.; Maier, M.K.; du Pasquier, L.; Ravens, I.; Dreyer, L.; Burger, R.; Gramatzki, M.; Schwinzer, R.; et al. CD96 interaction with CD155 via its first Ig-like domain is modulated by alternative splicing or mutations in distal Ig-like domains. J. Biol. Chem. 2009, 284, 2235–2244. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhang, H.; Li, M.; Hu, D.; Li, C.; Ge, B.; Jin, B.; Fan, Z. Recruitment of Grb2 and SHIP1 by the ITT-like motif of TIGIT suppresses granule polarization and cytotoxicity of NK cells. Cell Death Diff. 2013, 20, 456–464. [Google Scholar] [CrossRef]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Xia, P.; Du, Y.; Liu, S.; Huang, G.; Chen, J.; Zhang, H.; Hou, N.; Cheng, X.; Zhou, L.; et al. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (CD155) ligand engagement suppresses interferongamma production of natural killer cells via beta-arrestin 2-mediated negative signaling. J. Biol. Chem. 2014, 289, 17647–17657. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Sun, H.; Huang, Q.; Huang, M.; Wen, H.; Lin, R.; Zheng, M.; Qu, K.; Li, K.; Wei, H.; Xiao, W.; et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology 2019, 70, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Blake, S.J.; Dougall, W.C.; Miles, J.J.; Teng, M.W.; Smyth, M.J. Molecular Pathways: Targeting CD96 and TIGIT for Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 5183–5188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; Hu, Y.; Lu, C.; Li, J.; Liu, W.; He, Y.; Wang, P.; Cheng, C.; Hu, Y.U.; Huang, S.; et al. Cluster of differentiation 96 as a leukemia stem cell-specific marker and a factor for prognosis evaluation in leukemia. Mol. Clin. Oncol. 2015, 3, 833–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Xu, P.; Yao, D.; Chen, X.; Dai, H. CD33, CD96 and death associated protein kinase (DAPK) expression are associated with the survival rate and/or response to the chemotherapy in the patients with acute myeloid leukemia (AML). Med. Sci. Monit. 2017, 23, 1725–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2019, in press. [Google Scholar] [CrossRef] [Green Version]
- Degos, C.; Heinemann, M.; Barrou, J.; Boucherit, N.; Lambaudie, E.; Savina, A.; Gorvel, L.; Olive, D. Endometrial tumor microenvironment alters human NK cell recruitment, and resident NK cell phenotype and function. Front. Immunol. 2019, 10, 877. [Google Scholar] [CrossRef]
- Tang, W.; Pan, X.; Han, D.; Rong, D.; Zhang, M.; Yang, L.; Ying, J.; Guan, H.; Chen, Z.; Wang, X. Clinical significance of CD8(+) T cell immunoreceptor with Ig and ITIM domains(+) in locally advanced gastric cancer treated with SOX regimen after D2 gastrectomy. Oncoimmunology 2019, 8, e1593807. [Google Scholar] [CrossRef] [Green Version]
- Zingoni, A.; Fionda, C.; Borrelli, C.; Cippitelli, M.; Santoni, A.; Soriani, A. Natural Killer Cell Response to Chemotherapy-Stressed Cancer Cells: Role in Tumor Immunosurveillance. Front. Immunol. 2017, 8, 1194. [Google Scholar] [CrossRef] [Green Version]
- Fionda, C.; Stabile, H.; Molfetta, R.; Soriani, A.; Bernardini, G.; Zingoni, A.; Gismondi, A.; Paolini, R.; Cippitelli, M.; Santoni, A. Translating the anti-myeloma activity of Natural Killer cells into clinical application. Cancer Treat. Rev. 2018, 70, 255–264. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Molfetta, R.; Zitti, B.; Lecce, M.; Milito, N.D.; Stabile, H.; Fionda, C.; Cippitelli, M.; Gismondi, A.; Santoni, A.; Paolini, R. CD155: A Multi-Functional Molecule in Tumor Progression. Int. J. Mol. Sci. 2020, 21, 922. https://doi.org/10.3390/ijms21030922
Molfetta R, Zitti B, Lecce M, Milito ND, Stabile H, Fionda C, Cippitelli M, Gismondi A, Santoni A, Paolini R. CD155: A Multi-Functional Molecule in Tumor Progression. International Journal of Molecular Sciences. 2020; 21(3):922. https://doi.org/10.3390/ijms21030922
Chicago/Turabian StyleMolfetta, Rosa, Beatrice Zitti, Mario Lecce, Nadia Domenica Milito, Helena Stabile, Cinzia Fionda, Marco Cippitelli, Angela Gismondi, Angela Santoni, and Rossella Paolini. 2020. "CD155: A Multi-Functional Molecule in Tumor Progression" International Journal of Molecular Sciences 21, no. 3: 922. https://doi.org/10.3390/ijms21030922