Suppression of Hepatocyte Nuclear Factor 4 α by Long-term Infection of Hepatitis B Virus Contributes to Tumor Cell Proliferation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
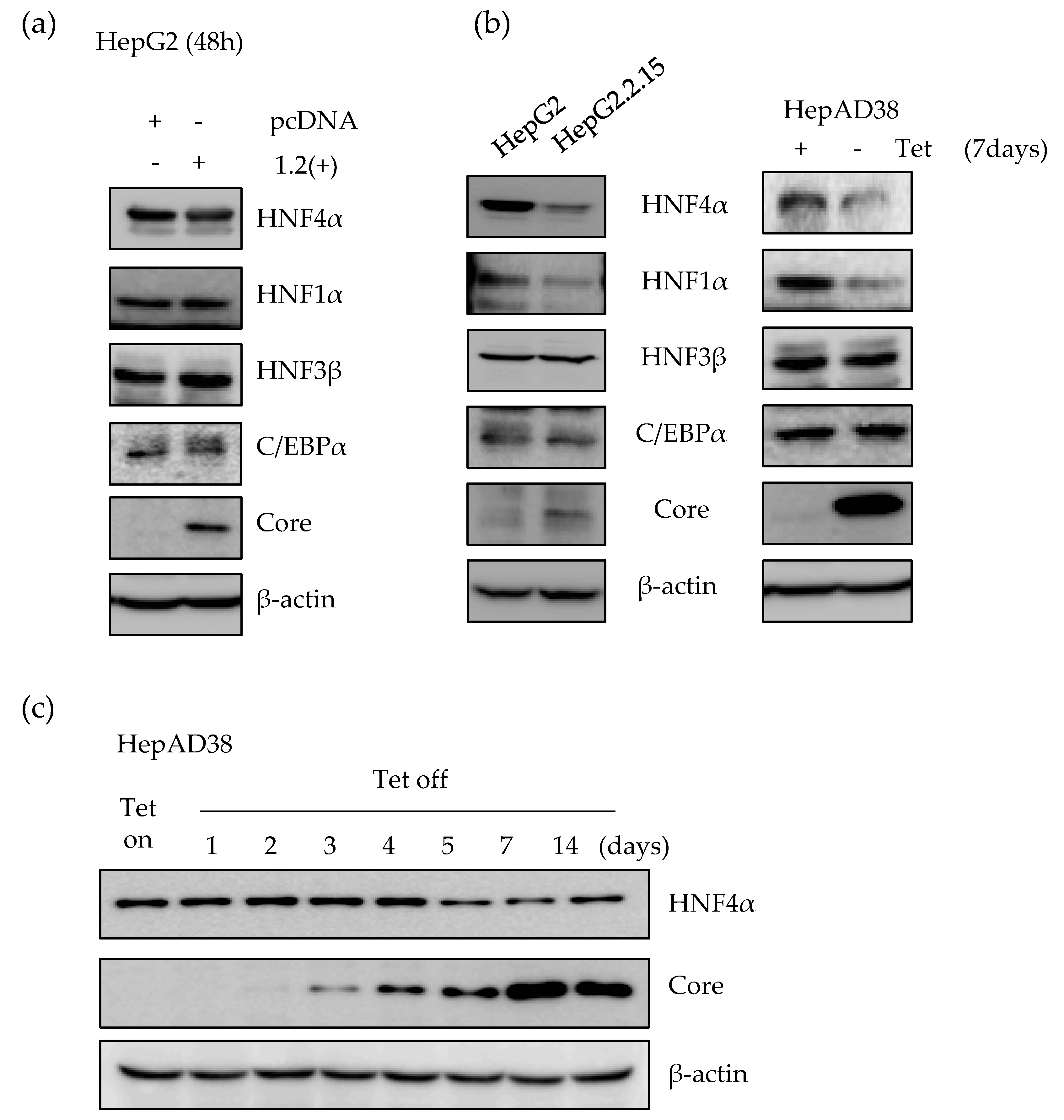
2.1. Expression of HNF4α Protein is Significantly Downregulated in Long-term Expression of HBV
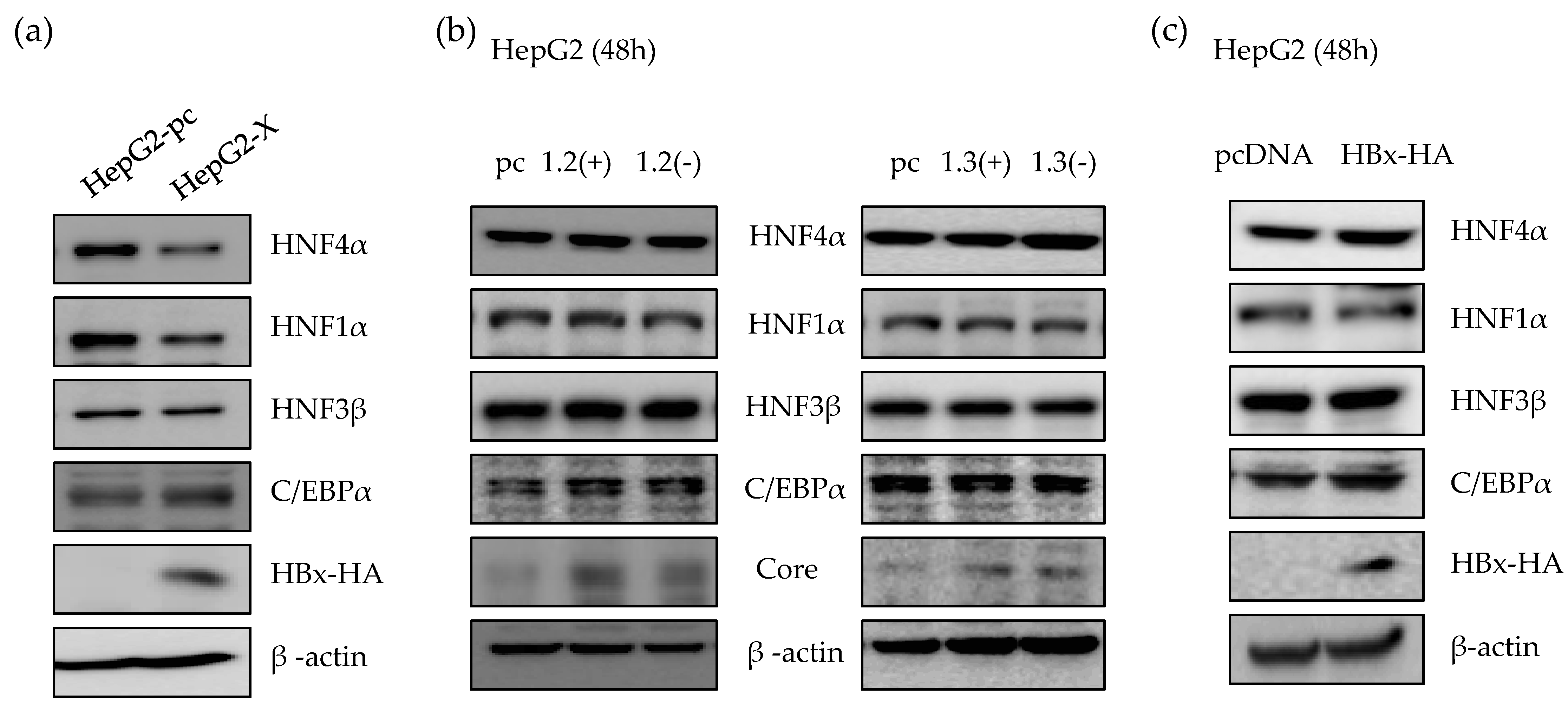
2.2. HBx Protein is Responsible for the Suppression of HNF4α in Long-term HBV Expression
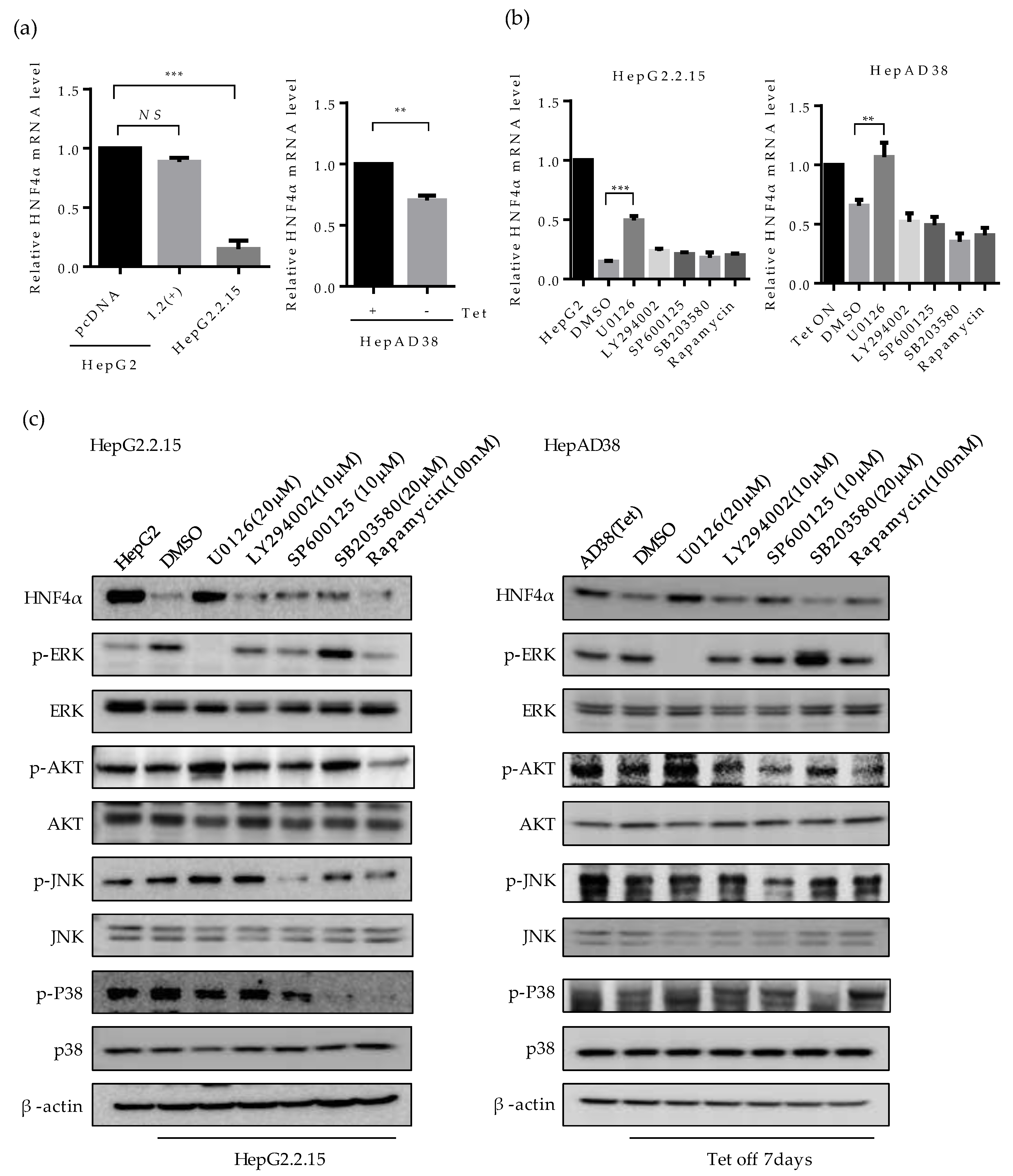
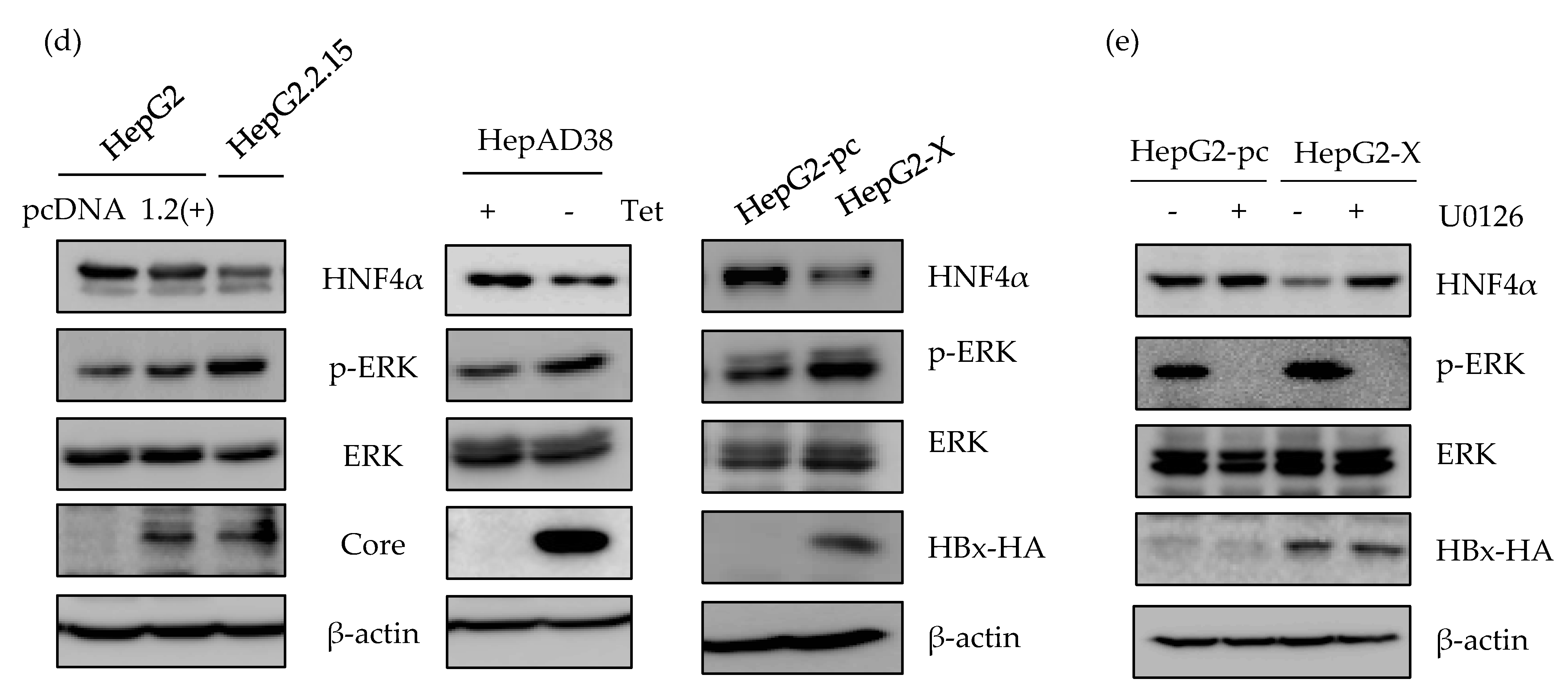
2.3. ERK Signaling Pathway is Associated with the Downregulation of HNF4α
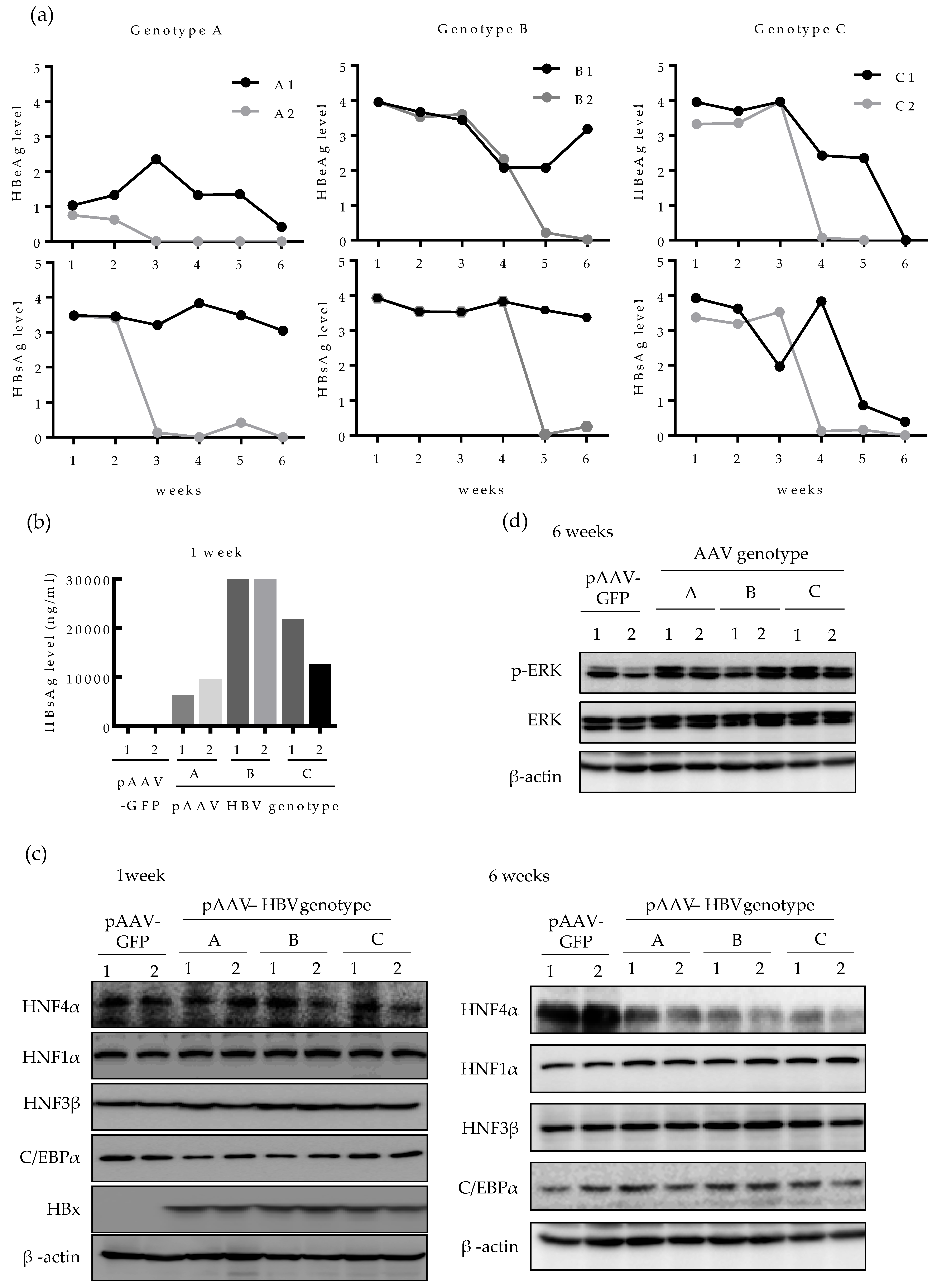
2.4. HNF4α Expression Is Suppressed in Long-term Expression of HBV in Mice
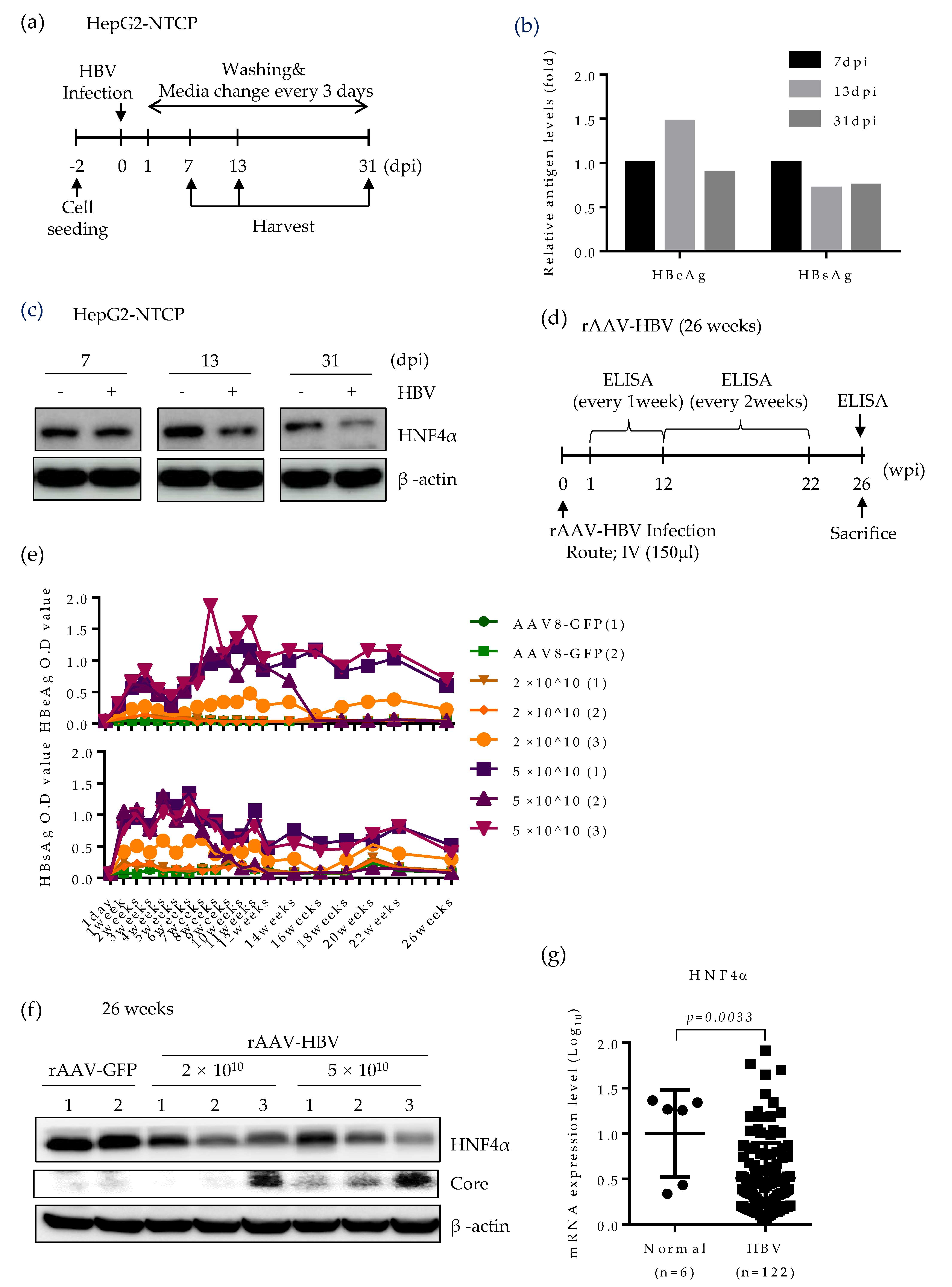
2.5. HNF4α Expression Is Suppressed during Long-term HBV Infection in vitro and in vivo
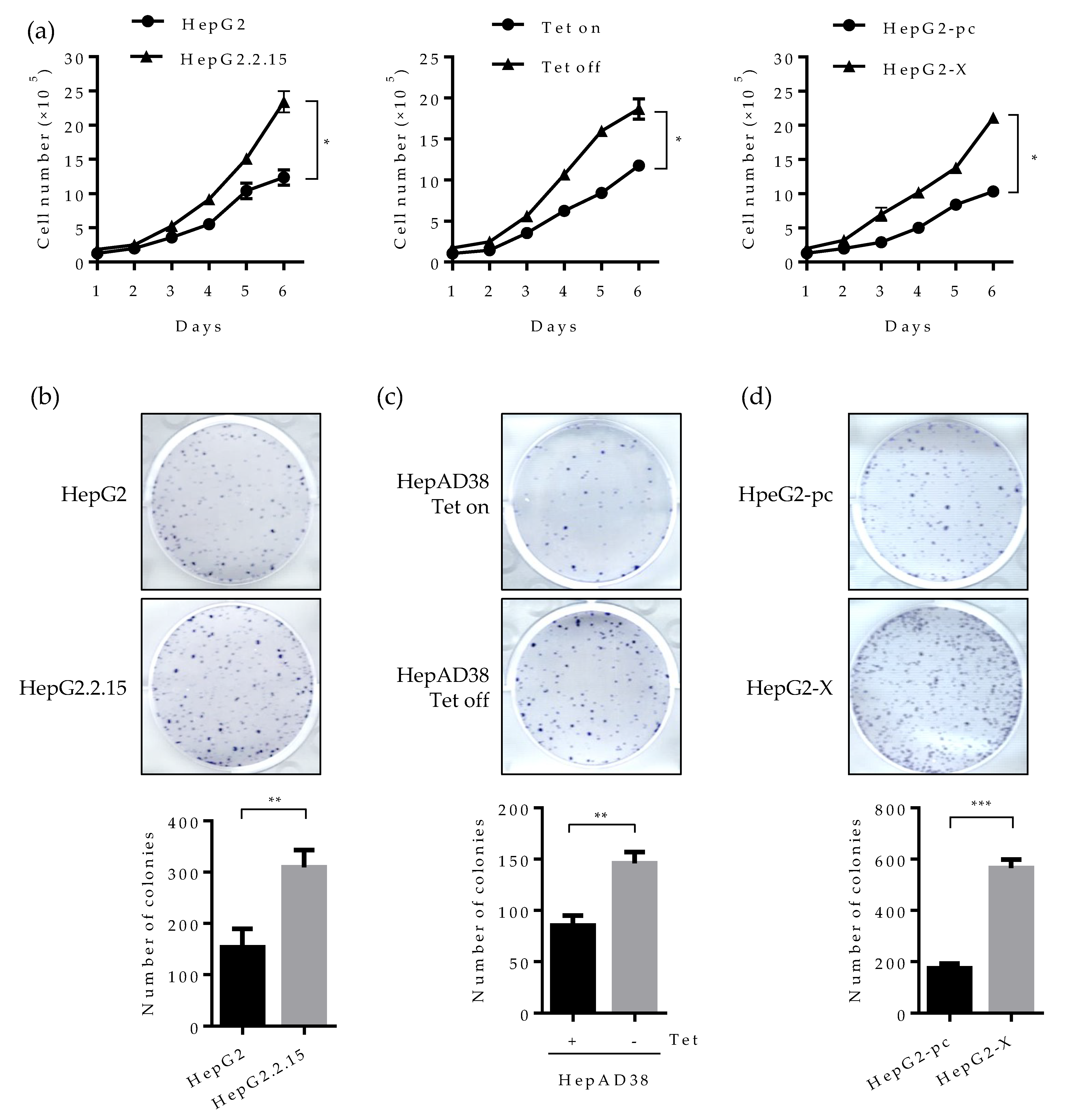
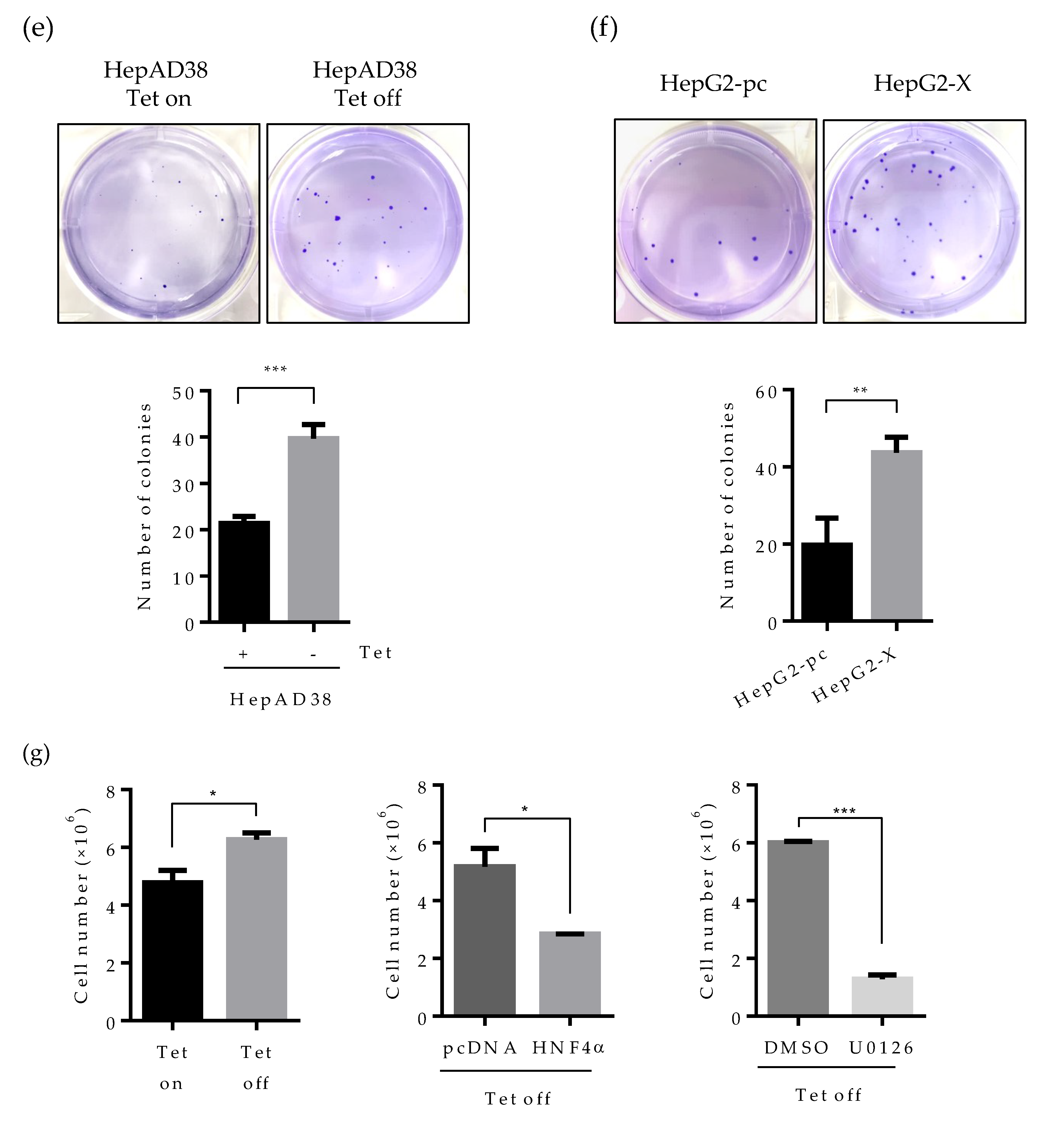
2.6. Suppression of HNF4α Enhances Cell Proliferation in the Sustained Presence of HBV
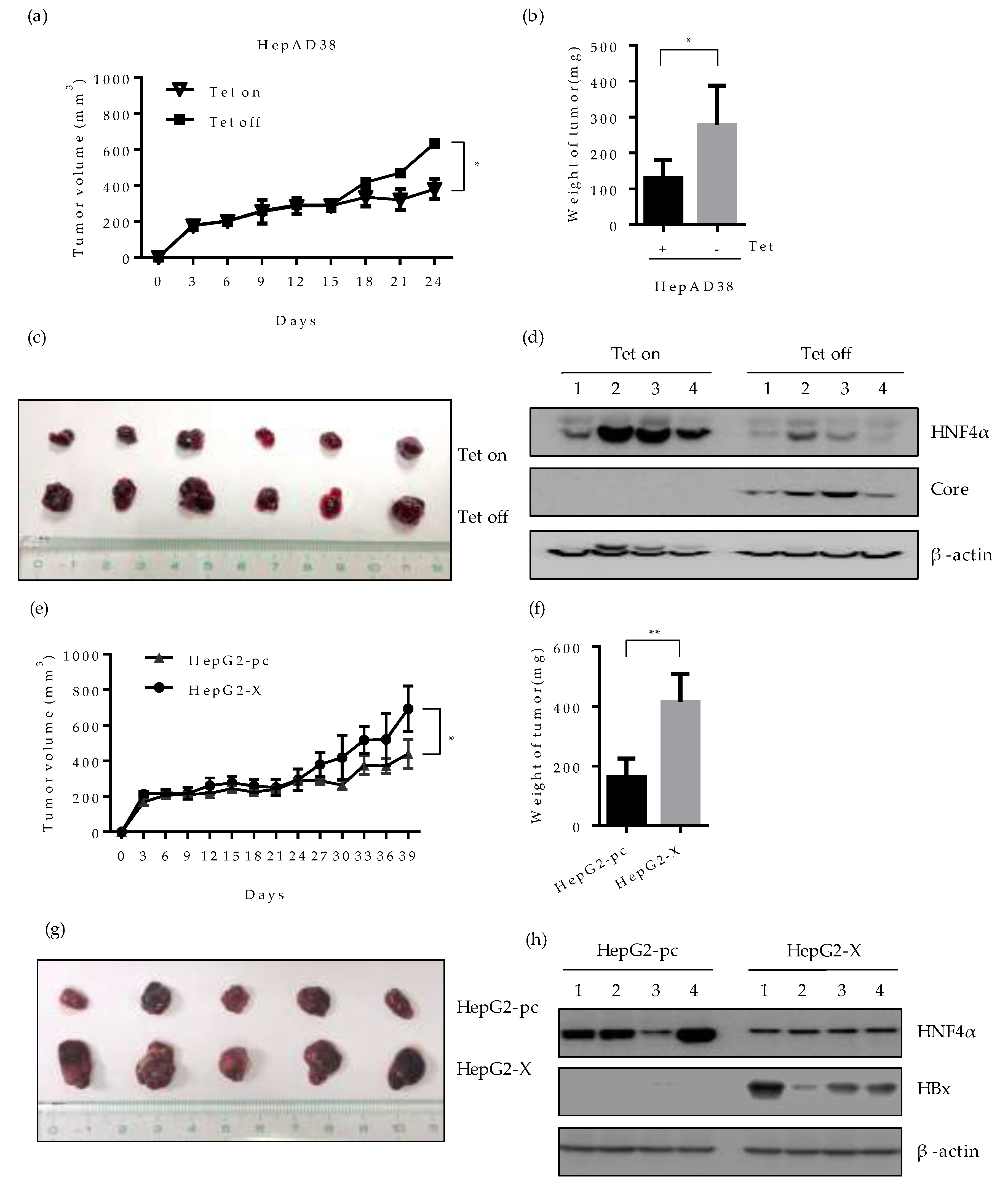
2.7. Effect of HBV-Induced Suppression of HNF4α on Tumor Formation and Growth in Xenograft Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasmid Construction
4.3. Animal Experiment
4.4. Transfection and Reagents
4.5. Virus Particle Production
4.6. Virus Infection
4.7. Western Blot
4.8. ELISA
4.9. Real-time Quantitative PCR (qRT-PCR)
4.10. Cell Proliferation and Colony-Forming Assay
4.11. Soft Agar Assay
4.12. Xenograft
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CHB | Chronic Hepatitis B Virus |
| dpi | Days post infection |
| Geq | Genome equivalent |
| HBeAg | HBV e antigen |
| HBsAg | HBV surface antigen |
| HBV | Hepatitis B virus |
| HBx | Hepatitis B virus X protein |
| HCC | Hepatocellular carcinoma |
| HNF1α | Hepatocyte nuclear factor 1 α |
| HNF3β | Hepatocyte nuclear factor 3 beta |
| HNF4α | Hepatocyte nuclear factor 4 α |
| IV | Intravenous |
| NTCP | Sodium-taurocholate cotransporting polypeptide |
| pAAV-HBV | Plasmid adeno associated virus-hepatitis B virus |
| rAAV-HBV | Recombinant adeno associated virus-hepatitis B virus |
| SC | Subcutaneous |
| Tet | Tetracycline |
| wpi | Weeks post infection |
References
- Ganem, D.; Prince, A.M. Hepatitis B virus infection--natural history and clinical consequences. N. Engl. J. Med. 2004, 350, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. Ca: A Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Hepatocellular carcinoma—United States, 2001–2006. MMWR 2010, 59, 517–520. [Google Scholar]
- Seeger, C.; Mason, W.S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef]
- De Meyer, S.; Gong, Z.J.; Suwandhi, W.; van Pelt, J.; Soumillion, A.; Yap, S.H. Organ and species specificity of hepatitis B virus (HBV) infection: A review of literature with a special reference to preferential attachment of HBV to human hepatocytes. J. Viral Hepat. 1997, 4, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Delgermaa, L.; Huang, F.; Oishi, N.; Liu, L.; He, F.; Zhao, L.; Murakami, S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J. Virol. 2005, 79, 5548–5556. [Google Scholar] [CrossRef] [PubMed]
- Brechot, C.; Kremsdorf, D.; Soussan, P.; Pineau, P.; Dejean, A.; Paterlini-Brechot, P.; Tiollais, P. Hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC): Molecular mechanisms and novel paradigms. Pathol. Biol. 2010, 58, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.A.; Lee, C. Hepatitis B virus X gene and hepatocarcinogenesis. J. Gastroenterol. 2011, 46, 974–990. [Google Scholar] [CrossRef]
- Hayhurst, G.P.; Lee, Y.H.; Lambert, G.; Ward, J.M.; Gonzalez, F.J. Hepatocyte nuclear factor 4α (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol. Cell. Biol. 2001, 21, 1393–1403. [Google Scholar] [CrossRef]
- Bonzo, J.A.; Ferry, C.H.; Matsubara, T.; Kim, J.H.; Gonzalez, F.J. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4α in adult mice. J. Biol. Chem. 2012, 287, 7345–7356. [Google Scholar] [CrossRef]
- Parviz, F.; Matullo, C.; Garrison, W.D.; Savatski, L.; Adamson, J.W.; Ning, G.; Kaestner, K.H.; Rossi, J.M.; Zaret, K.S.; Duncan, S.A. Hepatocyte nuclear factor 4α controls the development of a hepatic epithelium and liver morphogenesis. Nat. Genet. 2003, 34, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Battle, M.A.; Konopka, G.; Parviz, F.; Gaggl, A.L.; Yang, C.; Sladek, F.M.; Duncan, S.A. Hepatocyte nuclear factor 4α orchestrates expression of cell adhesion proteins during the epithelial transformation of the developing liver. Proc. Natl. Acad. Sci. USA 2006, 103, 8419–8424. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Jiang, S.; Hotta, H.; Takano, K.; Iwanari, H.; Sumi, K.; Daigo, K.; Ohashi, R.; Sugai, M.; Ikegame, C.; et al. Dysregulated expression of P1 and P2 promoter-driven hepatocyte nuclear factor-4α in the pathogenesis of human cancer. J. Pathol. 2006, 208, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Rawat, S.; Bouchard, M.J. The hepatitis B virus (HBV) HBx protein activates AKT to simultaneously regulate HBV replication and hepatocyte survival. J. Virol. 2015, 89, 999–1012. [Google Scholar] [CrossRef]
- Liu, H.; Lou, G.; Li, C.; Wang, X.; Cederbaum, A.I.; Gan, L.; Xie, B. HBx inhibits CYP2E1 gene expression via downregulating HNF4α in human hepatoma cells. PLoS ONE 2014, 9, e107913. [Google Scholar] [CrossRef]
- Chen, E.Q.; Sun, H.; Feng, P.; Gong, D.Y.; Liu, C.; Bai, L.; Yang, W.B.; Lei, X.Z.; Chen, L.Y.; Huang, F.J.; et al. Study of the expression levels of Hepatocyte nuclear factor 4 α and 3 beta in patients with different outcome of HBV infection. Virol. J. 2012, 9, 23. [Google Scholar] [CrossRef]
- Villanueva, A.; Newell, P.; Chiang, D.Y.; Friedman, S.L.; Llovet, J.M. Genomics and signaling pathways in hepatocellular carcinoma. Semin. Liver Dis. 2007, 27, 55–76. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Yu, P.; Ye, L.; Wang, H.; Du, G.; Zhang, J.; Zhang, J.; Tian, J. NSK-01105 inhibits proliferation and induces apoptosis of prostate cancer cells by blocking the Raf/MEK/ERK and PI3K/Akt/mTOR signal pathways. Tumour Biol. 2015, 36, 2143–2153. [Google Scholar] [CrossRef]
- Liao, B.; Zhou, H.; Liang, H.; Li, C. Regulation of ERK and AKT pathways by hepatitis B virus X protein via the Notch1 pathway in hepatocellular carcinoma. Int. J. Oncol. 2017, 51, 1449–1459. [Google Scholar] [CrossRef]
- Henkler, F.; Lopes, A.R.; Jones, M.; Koshy, R. Erk-independent partial activation of AP-1 sites by the hepatitis B virus HBx protein. J. Gen. Virol. 1998, 79, 2737–2742. [Google Scholar] [CrossRef] [PubMed]
- Nijhara, R.; Jana, S.S.; Goswami, S.K.; Rana, A.; Majumdar, S.S.; Kumar, V.; Sarkar, D.P. Sustained activation of mitogen-activated protein kinases and activator protein 1 by the hepatitis B virus X protein in mouse hepatocytes in vivo. J. Virol. 2001, 75, 10348–10358. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Park, E.S.; Lee, A.R.; Park, S.; Park, Y.K.; Ahn, S.H.; Kang, H.S.; Won, J.H.; Ha, Y.N.; Jae, B.; et al. Intracellular interleukin-32gamma mediates antiviral activity of cytokines against hepatitis B virus. Nat. Commun. 2018, 9, 3284. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241. [Google Scholar] [CrossRef]
- Walesky, C.; Apte, U. Role of hepatocyte nuclear factor 4α (HNF4α) in cell proliferation and cancer. Gene Expr. 2015, 16, 101–108. [Google Scholar] [CrossRef]
- Ali, A.; Abdel-Hafiz, H.; Suhail, M.; Al-Mars, A.; Zakaria, M.K.; Fatima, K.; Ahmad, S.; Azhar, E.; Chaudhary, A.; Qadri, I. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 10238–10248. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Wang, L.H.; Schneider, R.J. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef]
- Lee, A.R.; Lim, K.H.; Park, E.S.; Kim, D.H.; Park, Y.K.; Park, S.; Kim, D.S.; Shin, G.C.; Kang, H.S.; Won, J.; et al. Multiple Functions of Cellular FLIP Are Essential for Replication of Hepatitis B Virus. J. Virol. 2018, 92, e00339-18. [Google Scholar] [CrossRef]
- Slagle, B.L.; Bouchard, M.J. Hepatitis B Virus X and Regulation of Viral Gene Expression. Cold Spring Harb. Perspect. Med. 2016, 6, a021402. [Google Scholar] [CrossRef]
- Slagle, B.L.; Bouchard, M.J. Role of HBx in hepatitis B virus persistence and its therapeutic implications. Curr. Opin. Virol. 2018, 30, 32–38. [Google Scholar] [CrossRef]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Lin, Y.; Yin, C.; Zhang, X.; Ning, B.F.; Zhang, Q.; Zhang, J.P.; Qiu, L.; Qin, X.R.; Chen, Y.X.; et al. Recombinant adenovirus carrying the hepatocyte nuclear factor-1α gene inhibits hepatocellular carcinoma xenograft growth in mice. Hepatology 2011, 54, 2036–2047. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Wang, Z.; Chen, H.; Zhang, L.; Zhuo, F.; Yang, Q. Serum from Chronic Hepatitis B Patients Promotes Growth and Proliferation via the IGF-II/IGF-IR/MEK/ERK Signaling Pathway in Hepatocellular Carcinoma Cells. Cell. Physiol. Biochem. 2018, 47, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.; Gong, J.; Tian, D.; Wang, Z. Hepatitis B Virus X Protein Induces SATB1 Expression through Activation of ERK and p38MAPK Pathways to Suppress Anoikis. Dig. Dis. Sci. 2019, 64, 3203–3214. [Google Scholar] [CrossRef] [PubMed]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nature reviews. Cancer 2006, 6, 674–687. [Google Scholar] [PubMed]
- Yan, S.Y.; Fan, J.G.; Qio, L. Hepatitis B Virus (HBV) Infection and Hepatocellular Carcinoma- New Insights for an Old Topic. Curr. Cancer Drug Targets 2017, 17, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Muralidharan, P.; Raj, J.P. Update in global trends and aetiology of hepatocellular carcinoma. Contemp. Oncol. 2018, 22, 141–150. [Google Scholar] [CrossRef]
- Ko, C.; Lee, S.; Windisch, M.P.; Ryu, W.S. DDX3 DEAD-box RNA helicase is a host factor that restricts hepatitis B virus replication at the transcriptional level. J. Virol. 2014, 88, 13689–13698. [Google Scholar] [CrossRef]
- Lim, W.; Kwon, S.H.; Cho, H.; Kim, S.; Lee, S.; Ryu, W.S.; Cho, H. HBx targeting to mitochondria and ROS generation are necessary but insufficient for HBV-induced cyclooxygenase-2 expression. J. Mol. Med. 2010, 88, 359–369. [Google Scholar] [CrossRef]
- Huang, L.R.; Wu, H.L.; Chen, P.J.; Chen, D.S. An immunocompetent mouse model for the tolerance of human chronic hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2006, 103, 17862–17867. [Google Scholar] [CrossRef]
- Dezhbord, M.; Lee, S.; Kim, W.; Seong, B.L.; Ryu, W.S. Characterization of the molecular events of covalently closed circular DNA synthesis in de novo Hepatitis B virus infection of human hepatoma cells. Antivir. Res. 2019, 163, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Park, E.S.; Kim, D.H.; Ahn, S.H.; Park, S.H.; Lee, A.R.; Park, S.; Kang, H.S.; Lee, J.H.; Kim, J.M.; et al. Cleaved c-FLIP mediates the antiviral effect of TNF-α against hepatitis B virus by dysregulating hepatocyte nuclear factors. J. Hepatol. 2016, 64, 268–277. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Ha, Y.N.; Dezhbord, M.; Lee, A.R.; Park, E.-S.; Park, Y.K.; Won, J.; Kim, N.Y.; Choo, S.Y.; Shin, J.J.; et al. Suppression of Hepatocyte Nuclear Factor 4 α by Long-term Infection of Hepatitis B Virus Contributes to Tumor Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 948. https://doi.org/10.3390/ijms21030948
Park S, Ha YN, Dezhbord M, Lee AR, Park E-S, Park YK, Won J, Kim NY, Choo SY, Shin JJ, et al. Suppression of Hepatocyte Nuclear Factor 4 α by Long-term Infection of Hepatitis B Virus Contributes to Tumor Cell Proliferation. International Journal of Molecular Sciences. 2020; 21(3):948. https://doi.org/10.3390/ijms21030948
Chicago/Turabian StylePark, Soree, Yea Na Ha, Mehrangiz Dezhbord, Ah Ram Lee, Eun-Sook Park, Yong Kwang Park, Juhee Won, Na Yeon Kim, Soo Yeun Choo, Jae Jin Shin, and et al. 2020. "Suppression of Hepatocyte Nuclear Factor 4 α by Long-term Infection of Hepatitis B Virus Contributes to Tumor Cell Proliferation" International Journal of Molecular Sciences 21, no. 3: 948. https://doi.org/10.3390/ijms21030948
APA StylePark, S., Ha, Y. N., Dezhbord, M., Lee, A. R., Park, E.-S., Park, Y. K., Won, J., Kim, N. Y., Choo, S. Y., Shin, J. J., Ahn, C. H., & Kim, K.-H. (2020). Suppression of Hepatocyte Nuclear Factor 4 α by Long-term Infection of Hepatitis B Virus Contributes to Tumor Cell Proliferation. International Journal of Molecular Sciences, 21(3), 948. https://doi.org/10.3390/ijms21030948