Contributions of Myosin Light Chain Kinase to Regulation of Epithelial Paracellular Permeability and Mucosal Homeostasis

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Structure of Epithelial Intercellular Junctions
2. The Paracellular “Shunt” Pathway
3. MLCK, ZO-1, and Occludin Regulate the Leak Pathway
3.1. MLCK Regulates Leak Pathway Permeability
3.2. MLCK Regulates Tight Junction Protein Interactions and Structure
3.3. MLCK Activation Triggers Tight-Junction Protein Endocytosis
3.4. Interactions Mediated by the Occludin OCEL Domain Regulate Leak Pathway Barrier Function
3.5. MLCK-Induced Occludin Endocytosis Requires ZO-1 Interactions with the Occludin OCEL Domain
4. Regulation of MLCK Expression and Localization
4.1. Regulation of MLCK Transcription
4.2. MLCK Expression in Chronic Intestinal Disease
4.3. Enzymatic MLCK Inhibition is Not Feasible as a Therapeutic Intervention
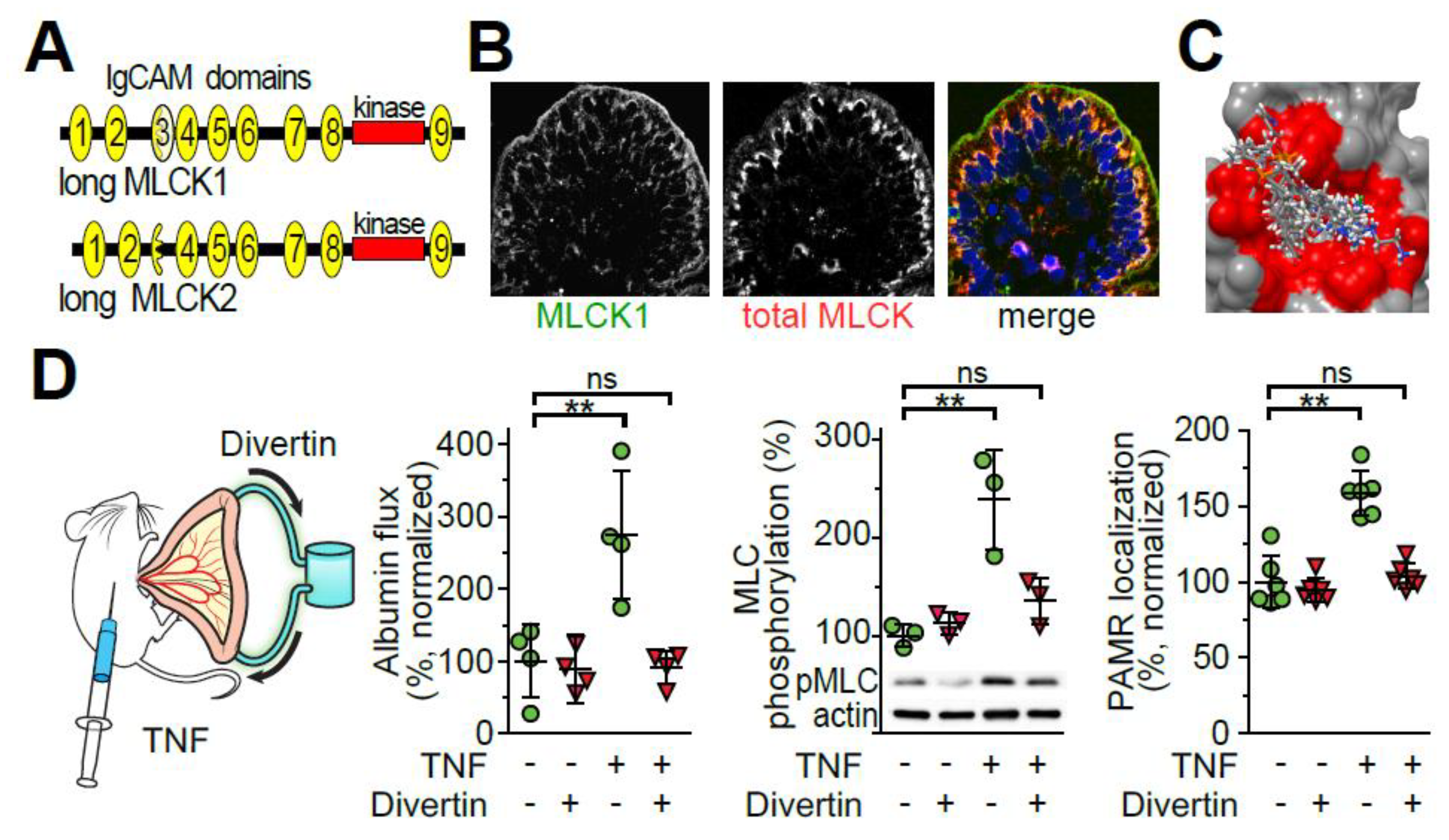
4.4. TNF Induces IgCAM3-Mediated Long MLCK1 Recruitment to the Perijunctional Actomyosin Ring
4.5. TNF Induces IgCAM3-Mediated Long MLCK1 Recruitment to the Perijunctional Actomyosin Ring
5. Perspective and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Blasky, A.J.; Mangan, A.; Prekeris, R. Polarized protein transport and lumen formation during epithelial tissue morphogenesis. Annu. Rev. Cell Dev. Biol. 2015, 31, 575–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatasubramanian, J.; Ao, M.; Rao, M.C. Ion transport in the small intestine. Curr. Opin. Gastroenterol. 2010, 26, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Cereijido, M.; Contreras, R.G.; Shoshani, L.; Flores-Benitez, D.; Larre, I. Tight junction and polarity interaction in the transporting epithelial phenotype. Biochim. Biophys. Acta 2008, 1778, 770–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawley, S.W.; Mooseker, M.S.; Tyska, M.J. Shaping the intestinal brush border. J. Cell Biol. 2014, 207, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farquhar, M.; Palade, G. Junctional complexes in various epithelia. J. Cell Biol. 1963, 17, 375–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cereijido, M.; Robbins, E.S.; Dolan, W.J.; Rotunno, C.A.; Sabatini, D.D. Polarized monolayers formed by epithelial cells on a permeable and translucent support. J. Cell Biol. 1978, 77, 853–880. [Google Scholar] [CrossRef]
- Martinez-Palomo, A.; Meza, I.; Beaty, G.; Cereijido, M. Experimental modulation of occluding junctions in a cultured transporting epithelium. J. Cell Biol. 1980, 87, 736–745. [Google Scholar] [CrossRef]
- Otani, T.; Nguyen, T.P.; Tokuda, S.; Sugihara, K.; Sugawara, T.; Furuse, K.; Miura, T.; Ebnet, K.; Furuse, M. Claudins and JAM-A coordinately regulate tight junction formation and epithelial polarity. J. Cell Biol. 2019, 218, 3372–3396. [Google Scholar] [CrossRef] [Green Version]
- Nalle, S.C.; Zuo, L.; Ong, M.; Singh, G.; Worthylake, A.M.; Choi, W.; Manresa, M.C.; Southworth, A.P.; Edelblum, K.L.; Baker, G.J.; et al. Graft-versus-host disease propagation depends on increased intestinal epithelial tight junction permeability. J. Clin. Investig. 2019, 129, 902–914. [Google Scholar] [CrossRef] [Green Version]
- Kuo, W.T.; Shen, L.; Zuo, L.; Shashikanth, N.; Ong, M.; Wu, L.; Zha, J.; Edelblum, K.L.; Wang, Y.; Wang, Y.; et al. Inflammation-induced Occludin Downregulation Limits Epithelial Apoptosis by Suppressing Caspase-3 Expression. Gastroenterology 2019, 157, 1323–1337. [Google Scholar] [CrossRef] [Green Version]
- Kohno, T.; Konno, T.; Kojima, T. Role of Tricellular Tight Junction Protein Lipolysis-Stimulated Lipoprotein Receptor (LSR) in Cancer Cells. Int. J. Mol. Sci. 2019, 20, 3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camilleri, M. Leaky gut: Mechanisms, measurement and clinical implications in humans. Gut 2019, 68, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Brazil, J.C.; Quiros, M.; Nusrat, A.; Parkos, C.A. Innate immune cell-epithelial crosstalk during wound repair. J. Clin. Investig. 2019, 129, 2983–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, D.W. Barrier function of epithelia. Am. J. Physiol. 1981, 241, G275–G288. [Google Scholar] [CrossRef] [PubMed]
- Kottra, G.; Fromter, E. Functional properties of the paracellular pathway in some leaky epithelia. J. Exp. Biol. 1983, 106, 217–229. [Google Scholar] [PubMed]
- Overton, J.; Shoup, J. Fine Structure of Cell Surface Specializations in the Maturing Duodenal Mucosa of the Chick. J. Cell Biol. 1964, 21, 75–85. [Google Scholar] [CrossRef]
- Tsubouchi, S.; Leblond, C.P. Migration and turnover of entero-endocrine and caveolated cells in the epithelium of the descending colon, as shown by radioautography after continuous infusion of 3H-thymidine into mice. Am. J. Anat. 1979, 156, 431–451. [Google Scholar] [CrossRef]
- Harris, T.J.; Tepass, U. Adherens junctions: From molecules to morphogenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 502–514. [Google Scholar] [CrossRef]
- Pinheiro, D.; Bellaiche, Y. Mechanical Force-Driven Adherens Junction Remodeling and Epithelial Dynamics. Dev. Cell 2018, 47, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Tsukita, S.; Tsukita, S.; Nagafuchi, A.; Yonemura, S. Molecular linkage between cadherins and actin filaments in cell-cell adherens junctions. Curr. Opin. Cell Biol. 1992, 4, 834–839. [Google Scholar] [CrossRef]
- Brooke, M.A.; Nitoiu, D.; Kelsell, D.P. Cell-cell connectivity: Desmosomes and disease. J. Pathol. 2012, 226, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Capaldo, C.T.; Farkas, A.E.; Nusrat, A. Epithelial adhesive junctions. F1000prime Rep. 2014, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Nava, P.; Laukoetter, M.G.; Hopkins, A.M.; Laur, O.; Gerner-Smidt, K.; Green, K.J.; Parkos, C.A.; Nusrat, A. Desmoglein-2: A novel regulator of apoptosis in the intestinal epithelium. Mol. Biol. Cell 2007, 18, 4565–4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zen, K.; Babbin, B.A.; Liu, Y.; Whelan, J.B.; Nusrat, A.; Parkos, C.A. JAM-C is a component of desmosomes and a ligand for CD11b/CD18-mediated neutrophil transepithelial migration. Mol. Biol. Cell 2004, 15, 3926–3937. [Google Scholar] [CrossRef] [PubMed]
- Chalcroft, J.P.; Bullivant, S. An interpretation of liver cell membrane and junction structure based on observation of freeze-fracture replicas of both sides of the fracture. J. Cell Biol. 1970, 47, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Claude, P.; Goodenough, D.A. Fracture faces of zonulae occludentes from “tight” and “leaky” epithelia. J. Cell Biol. 1973, 58, 390–400. [Google Scholar] [CrossRef] [Green Version]
- Wade, J.B.; Karnovsky, M.J. The structure of the zonula occludens. A single fibril model based on freeze-fracture. J. Cell Biol. 1974, 60, 168–180. [Google Scholar] [CrossRef]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef]
- Furuse, M.; Sasaki, H.; Fujimoto, K.; Tsukita, S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J. Cell Biol. 1998, 143, 391–401. [Google Scholar] [CrossRef]
- Furuse, M.; Sasaki, H.; Tsukita, S. Manner of interaction of heterogeneous claudin species within and between tight junction strands. J. Cell Biol. 1999, 147, 891–903. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Tokumasu, R.; Kimura, H.; Tsukita, S. Role of claudin species-specific dynamics in reconstitution and remodeling of the zonula occludens. Mol. Biol. Cell 2011, 22, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.A.; Kelly, J.M.; McCormack, J.; Rogers, R.A.; Lai, J.; Schneeberger, E.E.; Lynch, R.D. Rapid reduction of MDCK cell cholesterol by methyl-beta-cyclodextrin alters steady state transepithelial electrical resistance. Eur. J. Cell Biol. 1999, 78, 473–484. [Google Scholar] [CrossRef]
- Nusrat, A.; Parkos, C.A.; Verkade, P.; Foley, C.S.; Liang, T.W.; Innis-Whitehouse, W.; Eastburn, K.K.; Madara, J.L. Tight junctions are membrane microdomains. J. Cell Sci. 2000, 113, 1771–1781. [Google Scholar]
- Nusrat, A.; von Eichel-Streiber, C.; Turner, J.R.; Verkade, P.; Madara, J.L.; Parkos, C.A. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect. Immun. 2001, 69, 1329–1336. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.R.; Rill, B.K.; Carlson, S.L.; Carnes, D.; Kerner, R.; Mrsny, R.J.; Madara, J.L. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am. J. Physiol. 1997, 273, C1378–C1385. [Google Scholar] [CrossRef]
- Berglund, J.J.; Riegler, M.; Zolotarevsky, Y.; Wenzl, E.; Turner, J.R. Regulation of human jejunal transmucosal resistance and MLC phosphorylation by Na(+)-glucose cotransport. Am. J. Physiol.- Gastrointest. Liver Physiol. 2001, 281, G1487–G1493. [Google Scholar] [CrossRef]
- Madara, J.L.; Pappenheimer, J.R. Structural basis for physiological regulation of paracellular pathways in intestinal epithelia. J. Membr. Biol. 1987, 100, 149–164. [Google Scholar] [CrossRef]
- Atisook, K.; Carlson, S.; Madara, J.L. Effects of phlorizin and sodium on glucose-elicited alterations of cell junctions in intestinal epithelia. Am. J. Physiol. 1990, 258, C77–C85. [Google Scholar] [CrossRef]
- Atisook, K.; Madara, J.L. An oligopeptide permeates intestinal tight junctions at glucose-elicited dilatations. Gastroenterology 1991, 100, 719–724. [Google Scholar] [CrossRef]
- Turner, J.R.; Cohen, D.E.; Mrsny, R.J.; Madara, J.L. Noninvasive in vivo analysis of human small intestinal paracellular absorption: Regulation by Na+-glucose cotransport. Dig. Dis. Sci. 2000, 45, 2122–2126. [Google Scholar] [CrossRef]
- Pappenheimer, J.R. Physiological regulation of transepithelial impedance in the intestinal mucosa of rats and hamsters. J. Membr. Biol. 1987, 100, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Pappenheimer, J.R.; Reiss, K.Z. Contribution of solvent drag through intercellular junctions to absorption of nutrients by the small intestine of the rat. J. Membr. Biol. 1987, 100, 123–136. [Google Scholar] [CrossRef]
- Pappenheimer, J.R. Role of pre-epithelial “unstirred” layers in absorption of nutrients from the human jejunum. J. Membr. Biol. 2001, 179, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Pappenheimer, J.R. On the coupling of membrane digestion with intestinal absorption of sugars and amino acids. Am. J. Physiol. 1993, 265, G409–G417. [Google Scholar] [CrossRef] [PubMed]
- Pappenheimer, J.R.; Dahl, C.E.; Karnovsky, M.L.; Maggio, J.E. Intestinal absorption and excretion of octapeptides composed of D amino acids. Proc. Natl. Acad. Sci. USA 1994, 91, 1942–1945. [Google Scholar] [CrossRef] [Green Version]
- Pappenheimer, J.R. Physiological regulation of epithelial junctions in intestinal epithelia. Acta Physiol. Scand. Suppl. 1988, 571, 43–51. [Google Scholar]
- Pappenheimer, J.R. Paracellular intestinal absorption of glucose, creatinine, and mannitol in normal animals: Relation to body size. Am. J. Physiol. 1990, 259, G290–G299. [Google Scholar] [CrossRef]
- Meddings, J.B.; Westergaard, H. Intestinal glucose transport using perfused rat jejunum in vivo: Model analysis and derivation of corrected kinetic constants. Clin. Sci. 1989, 76, 403–413. [Google Scholar] [CrossRef]
- Pei, L.; Solis, G.; Nguyen, M.T.; Kamat, N.; Magenheimer, L.; Zhuo, M.; Li, J.; Curry, J.; McDonough, A.A.; Fields, T.A.; et al. Paracellular epithelial sodium transport maximizes energy efficiency in the kidney. J. Clin. Investig. 2016, 126, 2509–2518. [Google Scholar] [CrossRef] [Green Version]
- Zolotarevsky, Y.; Hecht, G.; Koutsouris, A.; Gonzalez, D.E.; Quan, C.; Tom, J.; Mrsny, R.J.; Turner, J.R. A membrane-permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 2002, 123, 163–172. [Google Scholar] [CrossRef]
- Clayburgh, D.R.; Barrett, T.A.; Tang, Y.; Meddings, J.B.; Van Eldik, L.J.; Watterson, D.M.; Clarke, L.L.; Mrsny, R.J.; Turner, J.R. Epithelial myosin light chain kinase-dependent barrier dysfunction mediates T cell activation-induced diarrhea in vivo. J. Clin. Investig. 2005, 115, 2702–2715. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R. Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 2009, 9, 799–809. [Google Scholar] [CrossRef]
- Anderson, J.M.; Van Itallie, C.M. Physiology and function of the tight junction. Cold Spring Harb. Perspect Biol. 2009, 1, a002584. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Graham, W.V.; Wang, Y.; Witkowski, E.D.; Schwarz, B.T.; Turner, J.R. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am. J. Pathol. 2005, 166, 409–419. [Google Scholar] [CrossRef]
- Ma, T.Y.; Boivin, M.A.; Ye, D.; Pedram, A.; Said, H.M. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: Role of myosin light-chain kinase protein expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G422–G430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.R.; Liang, G.H.; Wang, Y.; Das, S.; Shen, L.; Yu, A.S.; Nelson, D.J.; Turner, J.R. Claudin-2-dependent paracellular channels are dynamically gated. eLife 2015, 4, e09906. [Google Scholar] [CrossRef]
- Yu, A.S.; Cheng, M.H.; Angelow, S.; Gunzel, D.; Kanzawa, S.A.; Schneeberger, E.E.; Fromm, M.; Coalson, R.D. Molecular basis for cation selectivity in claudin-2-based paracellular pores: Identification of an electrostatic interaction site. J. Gen. Physiol. 2009, 133, 111–127. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhuo, M.; Pei, L.; Rajagopal, M.; Yu, A.S. Comprehensive cysteine-scanning mutagenesis reveals Claudin-2 pore-lining residues with different intrapore locations. J. Biol. Chem. 2014, 289, 6475–6484. [Google Scholar] [CrossRef] [Green Version]
- Wada, M.; Tamura, A.; Takahashi, N.; Tsukita, S. Loss of claudins 2 and 15 from mice causes defects in paracellular Na+ flow and nutrient transport in gut and leads to death from malnutrition. Gastroenterology 2013, 144, 369–380. [Google Scholar] [CrossRef]
- Tamura, A.; Hayashi, H.; Imasato, M.; Yamazaki, Y.; Hagiwara, A.; Wada, M.; Noda, T.; Watanabe, M.; Suzuki, Y.; Tsukita, S. Loss of claudin-15, but not claudin-2, causes Na+ deficiency and glucose malabsorption in mouse small intestine. Gastroenterology 2011, 140, 913–923. [Google Scholar] [CrossRef]
- Suzuki, H.; Tani, K.; Fujiyoshi, Y. Crystal structures of claudins: Insights into their intermolecular interactions. Ann. N. Y. Acad. Sci. 2017, 1397, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, R.; Gunzel, D.; Theune, D.; Czichos, C.; Schulzke, J.D.; Fromm, M. Water channels and barriers formed by claudins. Ann. N. Y. Acad. Sci. 2017, 1397, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Hernandez, V.; Quiros, M.; Nusrat, A. Intestinal epithelial claudins: Expression and regulation in homeostasis and inflammation. Ann. N. Y. Acad. Sci. 2017, 1397, 66–79. [Google Scholar] [CrossRef]
- Muto, S. Physiological roles of claudins in kidney tubule paracellular transport. Am. J. Physiol. Renal Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R.; Black, E.D. NHE3-dependent cytoplasmic alkalinization is triggered by Na(+)-glucose cotransport in intestinal epithelia. Am. J. Physiol. Cell Physiol. 2001, 281, C1533–C1541. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Shiue, H.; Palkon, S.; Wang, Y.; Cullinan, P.; Burkhardt, J.K.; Musch, M.W.; Chang, E.B.; Turner, J.R. Ezrin regulates NHE3 translocation and activation after Na+-glucose cotransport. Proc. Natl. Acad. Sci. USA 2004, 101, 9485–9490. [Google Scholar] [CrossRef] [Green Version]
- Shiue, H.; Musch, M.W.; Wang, Y.; Chang, E.B.; Turner, J.R. Akt2 phosphorylates ezrin to trigger NHE3 translocation and activation. J. Biol. Chem. 2005, 280, 1688–1695. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Wang, Y.; Graham, W.V.; Su, L.; Musch, M.W.; Turner, J.R. MAPKAPK-2 is a critical signaling intermediate in NHE3 activation following Na+-glucose cotransport. J. Biol. Chem. 2006, 281, 24247–24253. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Murtazina, R.; Cha, B.; Chakraborty, M.; Sarker, R.; Chen, T.E.; Lin, Z.; Hogema, B.M.; de Jonge, H.R.; Seidler, U.; et al. D-glucose acts via sodium/glucose cotransporter 1 to increase NHE3 in mouse jejunal brush border by a Na+/H+ exchange regulatory factor 2-dependent process. Gastroenterology 2011, 140, 560–571. [Google Scholar] [CrossRef] [Green Version]
- Bohlen, H.G. Na+-induced intestinal interstitial hyperosmolality and vascular responses during absorptive hyperemia. Am. J. Physiol. 1982, 242, H785–H789. [Google Scholar] [CrossRef]
- Clayburgh, D.R.; Musch, M.W.; Leitges, M.; Fu, Y.X.; Turner, J.R. Coordinated epithelial NHE3 inhibition and barrier dysfunction are required for TNF-mediated diarrhea in vivo. J. Clin. Investig. 2006, 116, 2682–2694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuhan, R.; Koutsouris, A.; Savkovic, S.D.; Hecht, G. Enteropathogenic Escherichia coli-induced myosin light chain phosphorylation alters intestinal epithelial permeability. Gastroenterology 1997, 113, 1873–1882. [Google Scholar] [CrossRef]
- Hecht, G.; Pestic, L.; Nikcevic, G.; Koutsouris, A.; Tripuraneni, J.; Lorimer, D.D.; Nowak, G.; Guerriero, V., Jr.; Elson, E.L.; Lanerolle, P.D. Expression of the catalytic domain of myosin light chain kinase increases paracellular permeability. Am. J. Physiol. 1996, 271, C1678–C1684. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Black, E.D.; Witkowski, E.D.; Lencer, W.I.; Guerriero, V.; Schneeberger, E.E.; Turner, J.R. Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J. Cell Sci. 2006, 119, 2095–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.; Shen, L.; Clayburgh, D.R.; Nalle, S.C.; Sullivan, E.A.; Meddings, J.B.; Abraham, C.; Turner, J.R. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology 2009, 136, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Charpentier, B.; Hiesse, C.; Lantz, O.; Ferran, C.; Stephens, S.; O’Shaugnessy, D.; Bodmer, M.; Benoit, G.; Bach, J.F.; Chatenoud, L. Evidence that antihuman tumor necrosis factor monoclonal antibody prevents OKT3-induced acute syndrome. Transplantation 1992, 54, 997–1002. [Google Scholar] [CrossRef]
- Ferran, C.; Dy, M.; Merite, S.; Sheehan, K.; Schreiber, R.; Leboulenger, F.; Landais, P.; Bluestone, J.; Bach, J.F.; Chatenoud, L. Reduction of morbidity and cytokine release in anti-CD3 MoAb-treated mice by corticosteroids. Transplantation 1990, 50, 642–648. [Google Scholar] [CrossRef]
- Ferran, C.; Sheehan, K.; Dy, M.; Schreiber, R.; Merite, S.; Landais, P.; Noel, L.H.; Grau, G.; Bluestone, J.; Bach, J.F.; et al. Cytokine-related syndrome following injection of anti-CD3 monoclonal antibody: Further evidence for transient in vivo T cell activation. Eur. J. Immunol. 1990, 20, 509–515. [Google Scholar] [CrossRef]
- Musch, M.W.; Clarke, L.L.; Mamah, D.; Gawenis, L.R.; Zhang, Z.; Ellsworth, W.; Shalowitz, D.; Mittal, N.; Efthimiou, P.; Alnadjim, Z.; et al. T cell activation causes diarrhea by increasing intestinal permeability and inhibiting epithelial Na+/K+-ATPase. J. Clin. Investig. 2002, 110, 1739–1747. [Google Scholar] [CrossRef]
- Tang, Y.; Clayburgh, D.R.; Mittal, N.; Goretsky, T.; Dirisina, R.; Zhang, Z.; Kron, M.; Ivancic, D.; Katzman, R.B.; Grimm, G.; et al. Epithelial NF-kappaB enhances transmucosal fluid movement by altering tight junction protein composition after T cell activation. Am. J. Pathol. 2010, 176, 158–167. [Google Scholar] [CrossRef]
- Beutel, O.; Maraspini, R.; Pombo-Garcia, K.; Martin-Lemaitre, C.; Honigmann, A. Phase Separation of Zonula Occludens Proteins Drives Formation of Tight Junctions. Cell 2019, 179, 923–936.e911. [Google Scholar] [CrossRef]
- Madara, J.L.; Carlson, S.; Anderson, J.M. ZO-1 maintains its spatial distribution but dissociates from junctional fibrils during tight junction regulation. Am. J. Physiol. 1993, 264, C1096–C1101. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Weber, C.R.; Turner, J.R. The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J. Cell Biol. 2008, 181, 683–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Marchiando, A.M.; Weber, C.R.; Raleigh, D.R.; Wang, Y.; Shen, L.; Turner, J.R. MLCK-dependent exchange and actin binding region-dependent anchoring of ZO-1 regulate tight junction barrier function. Proc. Natl. Acad. Sci. USA 2010, 107, 8237–8241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchiando, A.M.; Shen, L.; Graham, W.V.; Weber, C.R.; Schwarz, B.T.; Austin, J.R., 2nd; Raleigh, D.R.; Guan, Y.; Watson, A.J.; Montrose, M.H.; et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J. Cell Biol. 2010, 189, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, M.; Furuse, M.; Sasaki, H.; Schulzke, J.D.; Fromm, M.; Takano, H.; Noda, T.; Tsukita, S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol. Biol. Cell 2000, 11, 4131–4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulzke, J.D.; Gitter, A.H.; Mankertz, J.; Spiegel, S.; Seidler, U.; Amasheh, S.; Saitou, M.; Tsukita, S.; Fromm, M. Epithelial transport and barrier function in occludin-deficient mice. Biochim. Biophys. Acta 2005, 1669, 34–42. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Turner, J.R. Actin depolymerization disrupts tight junctions via caveolae-mediated endocytosis. Mol. Biol. Cell 2005, 16, 3919–3936. [Google Scholar] [CrossRef] [Green Version]
- Buschmann, M.M.; Shen, L.; Rajapakse, H.; Raleigh, D.R.; Wang, Y.; Wang, Y.; Lingaraju, A.; Zha, J.; Abbott, E.; McAuley, E.M.; et al. Occludin OCEL-domain interactions are required for maintenance and regulation of the tight junction barrier to macromolecular flux. Mol. Biol. Cell 2013, 24, 3056–3068. [Google Scholar] [CrossRef]
- Schwarz, B.T.; Wang, F.; Shen, L.; Clayburgh, D.R.; Su, L.; Wang, Y.; Fu, Y.X.; Turner, J.R. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology 2007, 132, 2383–2394. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.S.; McCarthy, K.M.; Francis, S.A.; McCormack, J.M.; Lai, J.; Rogers, R.A.; Lynch, R.D.; Schneeberger, E.E. Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. Am. J. Physiol. Cell Physiol. 2005, 288, C1231–C1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, D.; Guo, S.; Al-Sadi, R.; Ma, T.Y. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology 2011, 141, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Cording, J.; Berg, J.; Kading, N.; Bellmann, C.; Tscheik, C.; Westphal, J.K.; Milatz, S.; Gunzel, D.; Wolburg, H.; Piontek, J.; et al. In tight junctions, claudins regulate the interactions between occludin, tricellulin and marvelD3, which, inversely, modulate claudin oligomerization. J. Cell Sci. 2013, 126, 554–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raleigh, D.R.; Marchiando, A.M.; Zhang, Y.; Shen, L.; Sasaki, H.; Wang, Y.; Long, M.; Turner, J.R. Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol. Biol. Cell 2010, 21, 1200–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steed, E.; Elbediwy, A.; Vacca, B.; Dupasquier, S.; Hemkemeyer, S.A.; Suddason, T.; Costa, A.C.; Beaudry, J.B.; Zihni, C.; Gallagher, E.; et al. MarvelD3 couples tight junctions to the MEKK1-JNK pathway to regulate cell behavior and survival. J. Cell Biol. 2014, 204, 821–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steed, E.; Rodrigues, N.T.; Balda, M.S.; Matter, K. Identification of MarvelD3 as a tight junction-associated transmembrane protein of the occludin family. BMC Cell Biol. 2009, 10, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, S.M.; Amasheh, S.; Richter, J.F.; Milatz, S.; Gunzel, D.; Westphal, J.K.; Huber, O.; Schulzke, J.D.; Fromm, M. Tricellulin forms a barrier to macromolecules in tricellular tight junctions without affecting ion permeability. Mol. Biol. Cell 2009, 20, 3713–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krug, S.M.; Bojarski, C.; Fromm, A.; Lee, I.M.; Dames, P.; Richter, J.F.; Turner, J.R.; Fromm, M.; Schulzke, J.D. Tricellulin is regulated via interleukin-13-receptor alpha2, affects macromolecule uptake, and is decreased in ulcerative colitis. Mucosal. Immunol. 2018, 11, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Van Itallie, C.M.; Fanning, A.S.; Holmes, J.; Anderson, J.M. Occludin is required for cytokine-induced regulation of tight junction barriers. J. Cell Sci. 2010, 123, 2844–2852. [Google Scholar] [CrossRef] [Green Version]
- Schwayer, C.; Shamipour, S.; Pranjic-Ferscha, K.; Schauer, A.; Balda, M.; Tada, M.; Matter, K.; Heisenberg, C.P. Mechanosensation of Tight Junctions Depends on ZO-1 Phase Separation and Flow. Cell 2019, 179, 937–952 e918. [Google Scholar] [CrossRef]
- Spadaro, D.; Le, S.; Laroche, T.; Mean, I.; Jond, L.; Yan, J.; Citi, S. Tension-Dependent Stretching Activates ZO-1 to Control the Junctional Localization of Its Interactors. Curr. Biol. 2017, 27, 3783–3795 e3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Schwarz, B.T.; Graham, W.V.; Wang, Y.; Su, L.; Clayburgh, D.R.; Abraham, C.; Turner, J.R. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology 2006, 131, 1153–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.; Nalle, S.C.; Shen, L.; Turner, E.S.; Singh, G.; Breskin, L.A.; Khramtsova, E.A.; Khramtsova, G.; Tsai, P.Y.; Fu, Y.X.; et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology 2013, 145, 407–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, D.; Ma, I.; Ma, T.Y. Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G496–G504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.Y.; Iwamoto, G.K.; Hoa, N.T.; Akotia, V.; Pedram, A.; Boivin, M.A.; Said, H.M. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G367–G376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, M.D.; Mooseker, M.S. Characterization of the enterocyte-like brush border cytoskeleton of the C2BBe clones of the human intestinal cell line, Caco-2. J. Cell Sci. 1992, 102, 581–600. [Google Scholar] [PubMed]
- Ye, D.; Ma, T.Y. Cellular and molecular mechanisms that mediate basal and tumour necrosis factor-alpha-induced regulation of myosin light chain kinase gene activity. J. Cell Mol. Med. 2008, 12, 1331–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, W.V.; Wang, F.; Clayburgh, D.R.; Cheng, J.X.; Yoon, B.; Wang, Y.; Lin, A.; Turner, J.R. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J. Biol. Chem. 2006, 281, 26205–26215. [Google Scholar] [CrossRef] [Green Version]
- Al-Sadi, R.; Guo, S.; Ye, D.; Dokladny, K.; Alhmoud, T.; Ereifej, L.; Said, H.M.; Ma, T.Y. Mechanism of IL-1beta modulation of intestinal epithelial barrier involves p38 kinase and activating transcription factor-2 activation. J. Immunol. 2013, 190, 6596–6606. [Google Scholar] [CrossRef] [Green Version]
- Al-Sadi, R.; Guo, S.; Ye, D.; Ma, T.Y. TNF-alpha Modulation of Intestinal Epithelial Tight Junction Barrier Is Regulated by ERK1/2 Activation of Elk-1. Am. J. Pathol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Blair, S.A.; Kane, S.V.; Clayburgh, D.R.; Turner, J.R. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Investig. 2006, 86, 191–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelblum, K.L.; Sharon, G.; Singh, G.; Odenwald, M.A.; Sailer, A.; Cao, S.; Ravens, S.; Thomsen, I.; El Bissati, K.; McLeod, R.; et al. The microbiome activates CD4 T-cell-mediated immunity to compensate for increased intestinal permeability. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, J.M.; Florian, P.; Shen, L.; Graham, W.V.; Tretiakova, M.S.; Gitter, A.H.; Mrsny, R.J.; Turner, J.R. Distinct temporal-spatial roles for rho kinase and myosin light chain kinase in epithelial purse-string wound closure. Gastroenterology 2005, 128, 987–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamada, M.; Perez, T.D.; Nelson, W.J.; Sheetz, M.P. Two distinct modes of myosin assembly and dynamics during epithelial wound closure. J. Cell Biol. 2007, 176, 27–33. [Google Scholar] [CrossRef]
- Bement, W.M.; Forscher, P.; Mooseker, M.S. A novel cytoskeletal structure involved in purse string wound closure and cell polarity maintenance. J. Cell Biol. 1993, 121, 565–578. [Google Scholar] [CrossRef]
- Lechuga, S.; Ivanov, A.I. Disruption of the epithelial barrier during intestinal inflammation: Quest for new molecules and mechanisms. Biochim. Biophys. Acta 2017, 1864, 1183–1194. [Google Scholar] [CrossRef]
- Isobe, K.; Raghuram, V.; Krishnan, L.; Chou, C.L.; Yang, C.R.; Knepper, M.A. CRISPR-Cas9/Phosphoproteomics Identifies Multiple Non-canonical Targets of Myosin Light Chain Kinase. Am. J. Physiol. Renal Physiol. 2020. [Google Scholar] [CrossRef]
- Kamm, K.E.; Stull, J.T. Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 2001, 276, 4527–4530. [Google Scholar] [CrossRef] [Green Version]
- Garcia, J.G.; Lazar, V.; Gilbert-McClain, L.I.; Gallagher, P.J.; Verin, A.D. Myosin light chain kinase in endothelium: Molecular cloning and regulation. Am. J. Respir. Cell Mol. Biol. 1997, 16, 489–494. [Google Scholar] [CrossRef]
- Khromov, A.S.; Wang, H.; Choudhury, N.; McDuffie, M.; Herring, B.P.; Nakamoto, R.; Owens, G.K.; Somlyo, A.P.; Somlyo, A.V. Smooth muscle of telokin-deficient mice exhibits increased sensitivity to Ca2+ and decreased cGMP-induced relaxation. Proc. Natl. Acad. Sci. USA 2006, 103, 2440–2445. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, N.; Khromov, A.S.; Somlyo, A.P.; Somlyo, A.V. Telokin mediates Ca2+-desensitization through activation of myosin phosphatase in phasic and tonic smooth muscle. J. Muscle Res. Cell Motil. 2004, 25, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Nieznanski, K.; Sobieszek, A. Telokin (kinase-related protein) modulates the oligomeric state of smooth-muscle myosin light-chain kinase and its interaction with myosin filaments. Biochem. J. 1997, 322, 65–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somlyo, A.V.; Wang, H.; Choudhury, N.; Khromov, A.S.; Majesky, M.; Owens, G.K.; Somlyo, A.P. Myosin light chain kinase knockout. J. Muscle Res. Cell Motil. 2004, 25, 241–242. [Google Scholar] [CrossRef] [PubMed]
- He, W.Q.; Peng, Y.J.; Zhang, W.C.; Lv, N.; Tang, J.; Chen, C.; Zhang, C.H.; Gao, S.; Chen, H.Q.; Zhi, G.; et al. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology 2008, 135, 610–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayburgh, D.R.; Rosen, S.; Witkowski, E.D.; Wang, F.; Blair, S.; Dudek, S.; Garcia, J.G.; Alverdy, J.C.; Turner, J.R. A differentiation-dependent splice variant of myosin light chain kinase, MLCK1, regulates epithelial tight junction permeability. J. Biol. Chem. 2004, 279, 55506–55513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birukov, K.G.; Csortos, C.; Marzilli, L.; Dudek, S.; Ma, S.F.; Bresnick, A.R.; Verin, A.D.; Cotter, R.J.; Garcia, J.G. Differential regulation of alternatively spliced endothelial cell myosin light chain kinase isoforms by p60(Src). J. Biol. Chem. 2001, 276, 8567–8573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, V.; Garcia, J.G. A single human myosin light chain kinase gene (MLCK.; MYLK). Genomics 1999, 57, 256–267. [Google Scholar] [CrossRef]
- Graham, W.V.; He, W.; Marchiando, A.M.; Zha, J.; Singh, G.; Li, H.S.; Biswas, A.; Ong, M.; Jiang, Z.H.; Choi, W.; et al. Intracellular MLCK1 diversion reverses barrier loss to restore mucosal homeostasis. Nat. Med. 2019, 25, 690–700. [Google Scholar] [CrossRef]
- Graham, W.V.; Magis, A.T.; Bailey, K.M.; Turner, J.R.; Ostrov, D.A. Crystallization and preliminary X-ray analysis of the human long myosin light-chain kinase 1-specific domain IgCAM3. Acta Crystallogr. Sect. F Struct Biol. Cryst Commun. 2011, 67, 221–223. [Google Scholar] [CrossRef]
- Madsen, K.L.; Malfair, D.; Gray, D.; Doyle, J.S.; Jewell, L.D.; Fedorak, R.N. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm. Bowel Dis. 1999, 5, 262–270. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, W.-Q.; Wang, J.; Sheng, J.-Y.; Zha, J.-M.; Graham, W.V.; Turner, J.R. Contributions of Myosin Light Chain Kinase to Regulation of Epithelial Paracellular Permeability and Mucosal Homeostasis. Int. J. Mol. Sci. 2020, 21, 993. https://doi.org/10.3390/ijms21030993
He W-Q, Wang J, Sheng J-Y, Zha J-M, Graham WV, Turner JR. Contributions of Myosin Light Chain Kinase to Regulation of Epithelial Paracellular Permeability and Mucosal Homeostasis. International Journal of Molecular Sciences. 2020; 21(3):993. https://doi.org/10.3390/ijms21030993
Chicago/Turabian StyleHe, Wei-Qi, Jing Wang, Jian-Ying Sheng, Juan-Min Zha, W. Vallen Graham, and Jerrold R. Turner. 2020. "Contributions of Myosin Light Chain Kinase to Regulation of Epithelial Paracellular Permeability and Mucosal Homeostasis" International Journal of Molecular Sciences 21, no. 3: 993. https://doi.org/10.3390/ijms21030993