Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence

Abstract
:1. Introduction
2. Metastasis and Tumor Repopulation Associated with Cancer Cells That Might Be Overlooked or Scored as “Dead” in Preclinical Assays
3. Therapy-Induced Cancer Cell Dormancy and Disease Relapse
3.1. Multinucleated/Polyploid Cancer Cells
3.2. Senescent Cancer Cells
3.3. Anti-Proliferative Drug-Tolerant “Persister” Cancer Cells
3.4. Adaptive Mutation in Persister Cells
- ➢
- Treatment of colon cancer cells with targeted therapies decreased their mismatch-repair (MMR) and homologous-recombination proficiency, resulting in their increased dependency on error-prone DNA repair.
- ➢
- Patient-derived tumor samples underwent a similar response, with samples from colorectal-cancer residual disease exhibiting downregulated MMR proteins.
- ➢
- Markers of DNA damage were elevated in colorectal-cancer cells treated with targeted therapies.
- ➢
- Levels of reactive oxygen species were concomitantly increased in these cells, which could account for the accumulation of DNA damage.
- ➢
- Treatment with targeted therapies triggered initiation of a stress-response program, leading to adaptive mutability and genetic instability, rather than simply exerting nonspecific effects that caused DNA damage.
- ➢
- As seen in adaptive mutability of microorganisms, the changes in DNA repair seen in cancer cells following treatment with targeted therapeutic drugs was transient and reverted to pre-treatment status once growth capability in the presence of the drugs was restored.
4. Therapy-Induced Cancer Cell Apoptosis and Disease Relapse
4.1. Role of Caspase-3 in Tumor Repopulation
4.2. Recovery of Apoptotic Cancer Cells from the Brink of Death
4.3. Apoptotic Threshold
5. The Role of Cancer Cell Fusion in Metastasis and Therapy Resistance
6. Can Cancer Recurrence Be Prevented?
7. Preclinical Studies of Anticancer Agents Often Generate Misleading and Clinically Irrelevant Information
7.1. The Standard In Vitro Colony Formation Assay Lacks Specificity
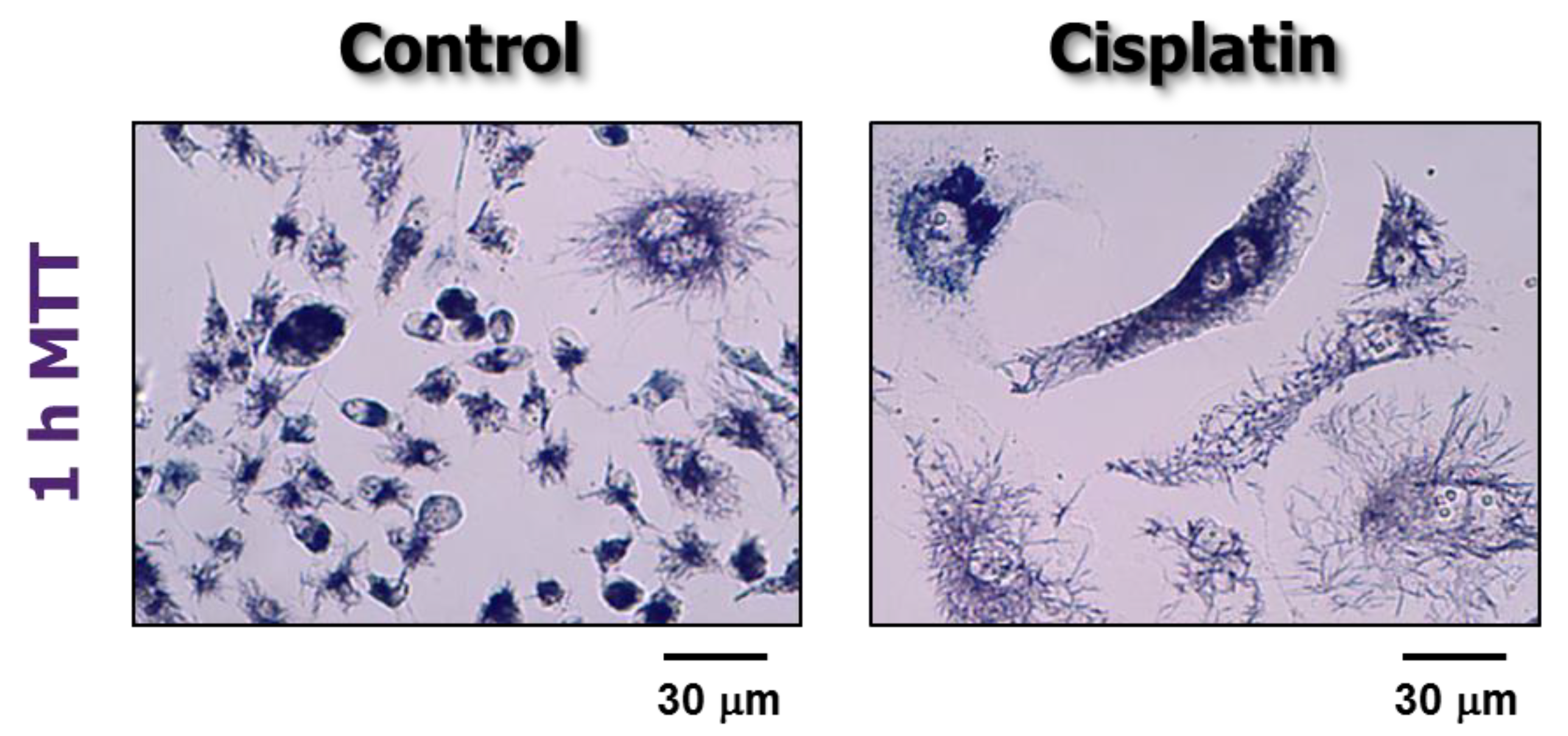
7.2. Multiwell Plate “Viability” and “Cytotoxicity” Assays Also Lack Specificity When Performed with Proliferating Cultures
7.3. Potential Use of Multiwell Plate Assays for Identifying Agents Capable of Killing Dormant Cancer Cells
8. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| NCI | National Cancer Institute |
| EMT | Epithelial to mesenchymal transition |
| PGCCs | Polyploid giant cancer cells |
| SIPS | Stress-induced premature senescence |
| CDK | Cyclin-dependent kinase |
| ATM | Ataxia telangiectasia mutated |
| SASP | Senescence-associated secretory phenotype |
| BRAF | Serine/threonine-protein kinase B-Raf |
| EGFR | epidermal growth factor receptor |
| MMR | Mismatch-repair |
| MOMP | Mitochondrial outer membrane permeabilization |
| APAF-1 | Apoptosis-activating factor-1 |
| DNase | Deoxyribonuclease |
| PS | Phosphatidylserine |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| NF-κB | Nuclear factor kappa B |
| STAT3 | Signal transducer and activator of transcription 3 |
| DFNA5 | Deafness, autosomal dominant 5 |
| CRM1 | Chromosomal region maintenance 1 |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide |
| IC50 | Half-maximum inhibitory concentration |
| XTT | 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilid |
| WST-1 | Water-soluble tetrazolium salt-1 |
References
- Weinberg, R.A. Coming full circle-from endless complexity to simplicity and back again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, L.V.; Shoemaker, R.H.; Paull, K.D.; Simon, R.M.; Tosini, S.; Skehan, P.; Scudiero, D.A.; Monks, A.; Boyd, M.R. Comparison of in vitro anticancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 1990, 82, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar]
- Scudiero, D.A.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar]
- Cell Viability—Promega Corporation. Available online: https://www.promega.ca/resources/guides/cell-biology/cell-viability/ (accessed on 10 February 2020).
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. In Assay Guidance Manual; Sittampalam, G.S., Ed.; Last Updated 2019; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004; pp. 295–319. [Google Scholar]
- Chen, D.L.; Engle, J.T.; Griffifin, E.A.; Miller, J.P.; Chu, W.; Zhou, D.; Mach, R.H. Imaging caspase-3 activation as a marker of apoptosis-targeted treatment response in cancer. Mol. Imaging Biol. 2015, 17, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Karamitopoulou, E.; Cioccari, L.; Jakob, S.; Vallan, C.; Schaffner, T.; Zimmermann, A.; Brunner, T. Active caspase 3 and DNA fragmentation as markers for apoptotic cell death in primary and metastatic liver tumours. Pathology 2007, 39, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Jonges, L.E.; Nagelkerke, J.F.; Ensink, N.G.; van der Velde, E.A.; Tollenaar, R.A.; Fleuren, G.J.; van de Velde, C.J.; Morreau, H.; Kuppen, P.J. Caspase-3 activity as a prognostic factor in colorectal carcinoma. Lab. Investig. 2001, 81, 681–688. [Google Scholar] [CrossRef] [Green Version]
- Niles, A.L.; Moravec, R.A.; Riss, T.L. Multiplex caspase activity and cytotoxicity assays. Methods Mol. Biol. 2008, 414, 151–162. [Google Scholar]
- Brown, J.M.; Wouters, B.G. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 1999, 59, 1391–1399. [Google Scholar] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Lauber, K.; Ernst, A.; Orth, M.; Herrmann, M.; Belka, C. Dying cell clearance and its impact on the outcome of tumor radiotherapy. Front. Oncol. 2012, 2, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansilla, S.; Priebe, W.; Portugal, J. Mitotic catastrophe results in cell death by caspase-dependent and caspase-independent mechanisms. Cell Cycle 2006, 5, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Li, J.; Jiang, E.; Wang, X.; Shangguan, A.J.; Zhang, L.; Yu, Z. Dormant Cells: The Original Cause of Tumor Recurrence and Metastasis. Cell Biochem. Biophys. 2015, 72, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.S.; Pandey, P.R.; Butti, R.; Radharani, N.N.V.; Roy, S.; Bhalara, S.R.; Gorain, M.; Kundu, G.C.; Kumar, D. The Biology and Therapeutic Implications of Tumor Dormancy and Reactivation. Front. Oncol. 2018, 8, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recasens, A.; Munoz, L. Targeting Cancer Cell Dormancy. Trends Pharmacol. Sci. 2019, 40, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef] [PubMed]
- Cancer Communications. The 150 most important questions in cancer research and clinical oncology series: Questions 94–101. Cancer Commun. 2018, 38, 69. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-H.; Torga, G.; Sun, Y.; Axelrod, R.; Pienta, K.J.; Sturm, J.C.; Austin, R.H. The role of heterogeneous environment and docetaxel gradient in the emergence of polyploid, mesenchymal and resistant prostate cancer cells. Clin. Exp. Metastasis 2019, 36, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Coward, J.; Harding, A. Size does matter: Why polyploid tumor cells are critical drug targets in the war on cancer. Front. Oncol. 2014, 4, 123. [Google Scholar] [CrossRef] [PubMed]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Kaur, E.; Rajendra, J.; Jadhav, S.; Shridhar, E.; Goda, J.S.; Moiyadi, A.; Dutt, S. Radiation-induced homotypic cell fusions of innately resistant glioblastoma cells mediate their sustained survival and recurrence. Carcinogenesis 2015, 36, 685–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Kumar, P.; Murray, D. Multinucleated giant cancer cells produced in response to ionizing radiation retain viability and replicate their genome. Int. J. Mol. Sci. 2017, 18, 360. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Impact of chemotherapeutic drugs on cancer cell proliferation, morphology and metabolic activity. J. Cancer Biol. Res. 2018, 6, 1118. [Google Scholar]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of polyploid/multinucleated giant cancer cells in metastasis and disease relapse following anticancer treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Viability assessment following anticancer treatment requires single-cell visualization. Cancers 2018, 10, 255. [Google Scholar] [CrossRef] [Green Version]
- Puck, T.T.; Marcus, P.I. Action of X-rays on mammalian cells. J. Exp. Med. 1956, 103, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Solari, F.; Domenget, C.; Gire, V.; Woods, C.; Lazarides, E.; Rousset, B.; Jurdic, P. Multinucleated cells can continuously generate mononucleated cells in the absence of mitosis: A study of cells of the avian osteoclast lineage. J. Cell Sci. 1995, 108, 3233–3241. [Google Scholar] [PubMed]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: A novel type of cell division in cancer. Cancer Biol. Ther. 2004, 3, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walen, K.H. Spontaneous cell transformation: Karyoplasts derived from multinucleated cells produce new cell growth in senescent human epithelial cell cultures. In Vitro Cell Dev. Biol. Anim. 2004, 40, 150–158. [Google Scholar] [CrossRef]
- Navolanic, P.M.; Akula, S.M.; McCubrey, J.A. Neosis and its potential role in cancer development and chemoresistance. Cancer Biol. Ther. 2004, 3, 219–320. [Google Scholar] [CrossRef] [Green Version]
- Rajaraman, R.; Rajaraman, M.M.; Rajaraman, S.R.; Guernsey, R.L. Neosis—A paradigm of self-renewal in cancer. Cell Biol. Int. 2005, 29, 1084–1097. [Google Scholar] [CrossRef]
- Walen, K.H. Budded karyoplasts from multinucleated fibroblast cells contain centrosomes and change their morphology to mitotic cells. Cell Biol. Int. 2005, 29, 1057–1065. [Google Scholar] [CrossRef]
- Rajaraman, R.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, S.R. Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Int. 2006, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Zhang, Q.; Wang, S.; Xie, S.; Fang, W.; Liu, Z.; Liu, J.; Yao, K. A fraction of CD133+ CNE2 cells is made of giant cancer cells with morphological evidence of asymmetric mitosis. J. Cancer 2015, 6, 1236–1244. [Google Scholar] [CrossRef] [Green Version]
- Esmatabadi, M.J.; Bakhshinejad, B.; Motlagh, F.M.; Babashah, S.; Sadeghizadeh, M. Therapeutic resistance and cancer recurrence mechanisms: Unfolding the story of tumour coming back. J. Biosci. 2016, 41, 497–506. [Google Scholar] [CrossRef]
- Lv, H.; Shi, Y.; Zhang, L.; Zhang, D.; Liu, G.; Yang, Z.; Li, Y.; Fei, F.; Zhang, S. Polyploid giant cancer cells with budding and the expression of cyclin E, S-phase kinase-associated protein 2, stathmin associated with the grading and metastasis in serous ovarian tumor. BMC Cancer 2014, 14, 576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Fei, F.; Zhang, D.; Yang, Z.; Wang, S.; Wang, X.; Wu, Z.; Wu, Q.; Zhang, S. The number of polyploid giant cancer cells and epithelial-mesenchymal transition-related proteins are associated with invasion and metastasis in human breast cancer. J. Exp. Clin. Cancer Res. 2015, 34, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Zhang, D.; Yang, Z.; Zhang, X. Tumor budding, micropapillary pattern, and polyploidy giant cancer cells in colorectal cancer: Current status and future prospects. Stem Cells Int. 2016, 2016, 4810734. [Google Scholar] [CrossRef] [Green Version]
- Mittal, K.; Donthamsetty, S.; Kaur, R.; Yang, C.; Gupta, M.V.; Reid, M.D.; Choi, D.H.; Rida, P.C.G.; Aneja, R. Multinucleated polyploidy drives resistance to Docetaxel chemotherapy in prostate Cancer. Br. J. Cancer 2017, 116, 1186–1194. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.A.; Cragg, M.S.; Fringes, B.; Sharakhov, I.; Illidge, T.M. Release of mitotic descendants by giant cells from irradiated Burkitt’s lymphoma cell lines. Cell Biol. Int. 2000, 24, 635–648. [Google Scholar] [CrossRef]
- Illidge, T.M.; Cragg, M.S.; Fringes, B.; Olive, P.; Erenpresia, J.A. Polyploid giant cells provide a survival mechanism of p53 mutant cells after DNA damage. Cell Biol. Int. 2000, 24, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Cragg, M.S. MOS, aneuploidy and the ploidy cycle of cancer cells. Oncogene 2010, 29, 5447–5451. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Ivanov, A.; Wheatley, P.S.; Kosmacek, E.A.; Ianzini, F.; Anisimov, A.P.; Mackey, M.; Davis, P.J.; Plakhins, G.; Illidge, T.M. Endopolyploidy in irradiated p53-defificient tumour cell lines: Persistence of cell division activity in giant cells expressing Aurora-B kinase. Cell Biol. Int. 2008, 32, 1044–1056. [Google Scholar] [CrossRef] [Green Version]
- Shu, Z.; Row, S.; Deng, W.M. Endoreplication: The good, the bad, and the ugly. Trends Cell Biol. 2018, 28, 465–474. [Google Scholar] [CrossRef]
- Díaz-Carballo, D.; Saka, S.; Klein, J.; Rennkamp, T.; Acikelli, A.H.; Malak, S.; Jastrow, H.; Wennemuth, G.; Tempfer, C.; Schmitz, I.; et al. A distinct oncogenerative multinucleated cancer cell serves as a source of stemness and tumor heterogeneity. Cancer Res. 2018, 78, 2318–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, F.; Zhang, M.; Li, B.; Zhao, L.; Wang, H.; Liu, L.; Li, Y.; Ding, P.; Gu, Y.; Zhang, X.; et al. Formation of polyploid giant cancer cells involves in the prognostic value of neoadjuvant chemoradiation in locally advanced rectal cancer. J. Oncol. 2019, 2019, 2316436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirois, I.; Aguilar-Mahecha, A.; Lafleur, J.; Fowler, E.; Vu, V.; Scriver, M.; Buchanan, M.; Chabot, C.; Ramanathan, A.; Balachandran, B.; et al. A unique morphological phenotype in chemoresistant triple-negative breast cancer reveals metabolic reprogramming and PLIN4 expression as a molecular vulnerability. Mol. Cancer Res. 2019, 17, 2492–2507. [Google Scholar] [CrossRef] [Green Version]
- Liu, J. The dualistic origin of human tumors. Semin. Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid Giant Cancer Cells (PGCCs): The Evil Roots of Cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “virgin birth”, polyploidy, and the origin of cancer. Oncoscience 2015, 2, 3–14. [Google Scholar] [CrossRef]
- Chen, H.; Lin, F.; Xing, K.; He, X. The reverse evolution from multicellularity to unicellularity during carcinogenesis. Nat. Commun. 2015, 6, 6367. [Google Scholar] [CrossRef]
- Masuda, M.; Wakasaki, T.; Toh, S. Stress-triggered atavistic reprogramming (STAR) addiction: Driving force behind head and neck cancer? Am. J. Cancer Res. 2016, 6, 1149–1166. [Google Scholar]
- Vincent, M. Resistance to cancer chemotherapy as an atavism? Bioessays 2016, 38, 1065. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pearson, R.B.; Papenfuss, A.T.; Goode, D.L. Altered interactions between unicellular and multicellular genes drive hallmarks of transformation in a diverse range of solid tumors. Proc. Natl. Acad. Sci. USA 2017, 114, 6406–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, F.; Ujvari, B.; Renaud, F.; Vincent, M. Cancer adaptations: Atavism, de novo selection, or something in between? Bioessays 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, Y.; Wang, Z. Evolution of oncogenic signatures of mutation hotspots in tyrosine kinases supports the atavistic hypothesis of cancer. Sci. Rep. 2018, 8, 8256. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V.; Vazquez-Martin, A.; Salmina, K.; Cragg, M.S. Stress-induced polyploidy shifts somatic cells towards a pro-tumourogenic unicellular gene transcription network. Cancer Hypotheses 2018, 1, 1–20. [Google Scholar]
- Niculescu, V.F. Carcinogenesis: Recent insights in protist stem cell biology lead to a better understanding of atavistic mechanisms implied in cancer development. MOJ Tumor Res. 2018, 1, 18–29. [Google Scholar]
- Mazzocca, A. The systemic–evolutionary theory of the origin of cancer (SETOC): A new interpretative model of cancer as a complex biological system. Int. J. Mol. Sci. 2019, 20, 4885. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, V.F. aCLS cancers: Genomic and epigenetic changes transform the cell of origin of cancer into a tumorigenic pathogen of unicellular organization and lifestyle. Gene 2020, 726, 144174. [Google Scholar] [CrossRef]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch, E.; Christov, K.; Roninson, I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999, 59, 3761–3767. [Google Scholar]
- Murray, D.; Mirzayans, R. Role of therapy-induced cellular senescence in tumor cells and its modifification in radiotherapy; the good, the bad and the ugly. J. Nucl. Med. Radiat. Ther. 2013, S6, 018. [Google Scholar]
- Jackson, J.G.; Pant, V.; Li, Q.; Chang, L.L.; Quintás-Cardama, A.; Garza, D.; Tavana, O.; Yang, P.; Manshouri, T.; Li, Y.; et al. p53-mediated senescence impairs the apoptotic response to chemotherapy and clinical outcome in breast cancer. Cancer Cell 2012, 21, 793–806. [Google Scholar] [CrossRef] [Green Version]
- Calvo Gonzalez, L.; Ghadaouia, S.; Martinez, A.; Rodier, F. Premature aging/senescence in cancer cells facing therapy: Good or bad? Biogerontology 2016, 17, 71–87. [Google Scholar] [CrossRef]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-step senescence-focused cancer therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.H.; Altschuler, S.J.; Wu, L.F. Patterns of early p21 dynamics determine proliferation-senescence cell fate after chemotherapy. Cell 2019, 178, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of wild-type p53 signaling in suppressing apoptosis in response to chemical genotoxic agents: Impact on chemotherapy outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crescenzi, E.; Palumbo, G.; de Boer, J.; Brady, H.J. Ataxia telangiectasia mutated and p21CIP1 modulate cell survival of drug-induced senescent tumor cells: Implications for chemotherapy. Clin. Cancer Res. 2008, 14, 1877–1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the senescence phenotype of cancer cells treated with chemotherapeutic agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Andrais, B.; Hansen, G.; Murray, D. Role of p16INK4A in replicative senescence and DNA damage-induced premature senescence in p53-defificient human cells. Biochem. Res. Int. 2012, 2012, 951574. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Morsbach, F.; Sander, D.; Gheorghiu, L.; Nanda, A.; Benes, C.; Kriegs, M.; Krause, M.; Dikomey, E.; Baumann, M.; et al. EGF receptor inhibition radiosensitizes NSCLC cells by inducing senescence in cells sustaining DNA double-strand breaks. Cancer Res. 2011, 71, 6261–6269. [Google Scholar] [CrossRef] [Green Version]
- McDermott, M.S.J.; Conlon, N.; Browne, B.C.; Szabo, A.; Synnott, N.C.; O’Brien, N.A.; Duffy, M.J.; Crown, J.; O’Donovan, N. HER2-targeted tyrosine kinase inhibitors cause therapy-induced-senescence in breast cancer cells. Cancers 2019, 11, 197. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davalos, A.R.; Coppé, J.P.; Campisi, J.; Desprez, P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sliwinska, M.A.; Mosieniak, G.; Wolanin, K.; Babik, A.; Piwocka, K.; Magalska, A.; Szczepanowska, J.; Fronk, J.; Sikora, E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech. Ageing Dev. 2009, 130, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy formation in doxorubicin-treated cancer cells can favor escape from senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [Green Version]
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D. Is senescence reversible? Curr. Drug Targets 2016, 17, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Vallette, F.M.; Olivier, C.; Lézot, F.; Oliver, L.; Cochonneau, D.; Lalier, L.; Cartron, P.F.; Heymann, D. Dormant, quiescent, tolerant and persister cells: Four synonyms for the same target in cancer. Biochem. Pharmacol. 2019, 162, 169–176. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, A.; Laureti, L.; Crussard, S.; Abida, H.; Rodríguez-Rojas, A.; Blázquez, J.; Baharoglu, Z.; Mazel, D.; Darfeuille, F.; Vogel, J.; et al. β-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity. Nat. Commun. 2013, 4, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponder, R.G.; Fonville, N.C.; Rosenberg, S.M. A switch from high-fidelity to errorprone DNA double-strand break repair underlies stress-induced mutation. Mol. Cell 2005, 19, 791–804. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, G.J.; Rosenberg, S.M. Adaptive mutations, mutator DNA polymerases and genetic change strategies of pathogens. Curr. Opin. Microbiol 2001, 4, 586–594. [Google Scholar] [CrossRef]
- Galhardo, R.S.; Hastings, P.J.; Rosenberg, S.M. Mutation as a stress response and the regulation of evolvability. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 399–435. [Google Scholar] [CrossRef] [Green Version]
- Foster, P.L. Adaptive mutation: Implications for evolution. BioEssays 2000, 22, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Taddei, F.; Radman, M.; Maynard-Smith, J.; Toupance, B.; Gouyon, P.H.; Godelle, B. Role of mutator alleles in adaptive evolution. Nature 1997, 387, 700–702. [Google Scholar] [CrossRef]
- Torkelson, J.; Harris, R.S.; Lombardo, M.J.; Nagendran, J.; Thulin, C.; Rosenberg, S.M. Genome-wide hypermutation in a subpopulation of stationary-phase cells underlies recombination-dependent adaptive mutation. EMBO J. 1997, 16, 3303–3311. [Google Scholar] [CrossRef]
- Rosche, W.A.; Foster, P.L. The role of transient hypermutators in adaptive mutation in Escherichia coli. Proc. Natl. Acad. Sci. USA 1999, 96, 6862–6867. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, G.J.; Harris, R.S.; Lee, P.L.; Rosenberg, S.M. The SOS response regulates adaptive mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 6646–6651. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, D.M.; Hastings, P.S.; Rosenberg, S.M. Stress-induced mutagenesis: Mplications in cancer and drug resistance. Annu. Rev. Cancer Biol. 2017, 1, 119–140. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, V.T.; Bindra, R.S.; Yuan, J.; Campisi, D.; Narayanan, L.; Jensen, R.; Giordano, F.; Johnson, R.S.; Rockwell, S.; Glazer, P.M. Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol. Cell. Biol. 2003, 23, 3265–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindra, R.S.; Glazer, P.M. Co-repression of mismatch repair gene expression by hypoxia in cancer cells: Role of the Myc/Max network. Cancer Lett. 2007, 252, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Weiss, R.H.; Murray, D. Spontaneous γH2AX foci in human solid tumor-derived cell lines in relation to p21WAF1 and WIP1 expression. Int. J. Mol. Sci. 2015, 16, 11609–11628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.M.; Hollville, E.; Martin, S.J. Measuring apoptosis by microscopy and flow cytometry. Methods 2013, 61, 90–97. [Google Scholar] [CrossRef]
- Zhao, R.; Kaakati, R.; Lee, A.K.; Liu, X.; Li, F.; Li, C.-Y. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev. 2018, 37, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.-N.; Crawford, J.C.; Heckmann, B.L.; Green, D.R. To the edge of cell death and back. FEBS J. 2019, 286, 430–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraguchi, M.; Torii, S.; Matsuzawa, S.I.; Xie, Z.; Kitada, S.; Krajewski, S.; Yoshida, H.; Mak, T.W.; Reed, J.C. Apoptotic protease activating factor 1 (Apaf-1)-independent cell death suppression by Bcl-2. J. Exp. Med. 2000, 191, 1709–1720. [Google Scholar] [CrossRef] [Green Version]
- Khosravi-Far, R. Death receptor signals to the mitochondria. Cancer Biol. Ther. 2004, 3, 1051–1057. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Ng, W.-L.; Huang, Q.; Liu, X.; Zimmerman, M.; Li, F.; Li, C.-Y. Molecular mechanisms involved in tumor repopulation after radiotherapy. Transl. Cancer Res. 2013, 2, 442–448. [Google Scholar]
- Cartwright, I.M.; Liu, X.; Zhou, M.; Li, F.; Li, C.-Y. Essential roles of Caspase-3 in facilitating Myc-induced genetic instability and carcinogenesis. eLife 2017, 6, e26371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakarla, R.; Hur, J.; Kim, Y.J.; Kim, J.; Chwae, Y.J. Apoptotic cell-derived exosomes: Messages from dying cells. Exp. Mol. Med. 2020, in press. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.L.; Huang, Q.; Liu, X.; Li, F.; Zimmerman, M.A.; Li, C.Y. Caspase 3 promotes surviving melanoma tumor cell growth after cytotoxic therapy. J. Investig. Dermatol. 2014, 134, 1686–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, L.; Meyer, M.; Fay, J.; Curry, S.; Bacon, O.; Duessmann, H.; John, K.; Boland, K.C.; McNamara, D.A.; Kay, E.W.; et al. Low levels of caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: Caspase-3 inhibition as a therapeutic approach. Cell Death Dis. 2016, 7, e2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Yu, Y.; He, S.; Cheng, J.; Gong, Y.; Zhang, Z.; Yang, X.; Xu, B.; Liu, X.; Li, C.Y.; et al. Dying glioma cells establish a proangiogenic microenvironment through a caspase 3 dependent mechanism. Cancer Lett. 2017, 385, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Liu, X.; Li, Z.; Huang, Q.; Li, F.; Li, C.Y. Caspase-3 regulates the migration, invasion and metastasis of colon cancer cells. Int. J. Cancer 2018, 143, 921–930. [Google Scholar] [CrossRef]
- Liu, X.; Li, F.; Huang, Q.; Zhang, Z.; Zhou, L.; Deng, Y.; Zhou, M.; Fleenor, D.E.; Wang, H.; Kastan, M.B.; et al. Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res. 2017, 27, 764–783. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Kim, J.M.; Kim, J.; Hur, J.; Park, S.; Kim, K.; Shin, H.-J.; Chwae, Y.-J. Molecular mechanisms of biogenesis of apoptotic exosome-like vesicles and their roles as damage-associated molecular patterns. Proc. Natl. Acad. Sci. USA 2018, 115, E11721–E11730. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.L.; Yuen, K.L.; Tang, H.M.; Fung, M.C. Reversibility of apoptosis in cancer cells. Br. J. Cancer 2009, 100, 118–122. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.M.; Tang, H.L. Anastasis: Recovery from the brink of cell death. R. Soc. Open Sci. 2018, 5, 180442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional ways to live and die: Cell death and survival in development, homeostasis, and disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Xie, X.; Wong, C.S.; Choi, Y.; Fung, M.C. HepG2 cells recovered from apoptosis show altered drug responses and invasiveness. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 293–300. [Google Scholar] [CrossRef]
- Tang, H.L.; Tang, H.M.; Hardwick, J.M.; Fung, M.C. Strategies for tracking anastasis, a cell survival phenomenon that reverses apoptosis. J. Vis. Exp. JoVE 2015, 96, e51964. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.M.; Talbot, C.C., Jr.; Fung, M.C.; Tang, H.L. Molecular signature of anastasis for reversal of apoptosis. F1000Research 2017, 6, 43. [Google Scholar] [CrossRef]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; So, C.; Lam, H.-M.; Fung, M.-C.; Tsang, S.-Y. Apoptosis reversal promotes cancer stem cell-like cell formation. Neoplasia 2018, 20, 295–303. [Google Scholar] [CrossRef]
- Xu, Y.; So, C.; Lam, H.M.; Fung, M.C.; Tsang, S.Y. Flow Cytometric detection of newly-formed breast cancer stem cell-like cells after apoptosis reversal. J. Vis. Exp. 2019, 143, e58642. [Google Scholar] [CrossRef] [Green Version]
- Seervi, M.; Sumi, S.; Chandrasekharan, A.; Sharma, A.K.; SanthoshKumar, T.R. Molecular profiling of anastatic cancer cells: Potential role of the nuclear export pathway. Cell Oncol. 2019, 42, 645–661. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Do multiwell plate high throughput assays measure loss of cell viability following exposure to genotoxic agents? Int. J. Mol. Sci. 2017, 18, 1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. The growing complexity of cancer cell response to DNA-damaging agents: Caspase 3 mediates cell death or survival? Int. J. Mol. Sci. 2016, 17, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, D.; Mirzayans, R. Non-linearities in the cellular response to ionizing radiation and the role of p53 therein. Int. J. Radiat. Biol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Pawelek, J.M. Fusion of bone marrow-derived cells with cancer cells: Metastasis as a secondary disease in cancer. Chin. J. Cancer 2014, 33, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clawson, G.A. Cancer. Fusion for moving. Science 2013, 342, 699–700. [Google Scholar] [CrossRef] [PubMed]
- Laberge, G.S.; Duvall, E.; Haedicke, K.; Pawelek, J. Leukocyte–Cancer Cell Fusion—Genesis of a deadly journey. Cells 2019, 8, 170. [Google Scholar] [CrossRef] [Green Version]
- Jiang, E.; Yan, T.; Xu, Z.; Shang, Z. Tumor Microenvironment and Cell Fusion. Biomed. Res. Int. 2019, 2019, 5013592. [Google Scholar] [CrossRef]
- Bastida-Ruiz, D.; Van Hoesen, K.; Cohen, M. The dark side of cell fusion. Int. J. Mol. Sci. 2016, 17, 638. [Google Scholar] [CrossRef] [Green Version]
- Platt, J.L.; Zhou, X.; Lefferts, A.R.; Cascalho, M. Cell fusion in the war on cancer: A perspective on the inception of malignancy. Int. J. Mol. Sci. 2016, 17, 1118. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, C.; Prabhu, P.; Juvale, K.; Suares, D.; Yc, M. Cancer cell fusion: A potential target to tackle drug-resistant and metastatic cancer cells. Drug Discov. Today 2019, 24, 1836–1844. [Google Scholar] [CrossRef]
- Clawson, G. The Fate of Fusions. Cells 2019, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gast, C.E.; Silk, A.D.; Zarour, L.; Riegler, L.; Burkhart, J.G.; Gustafson, K.T.; Parappilly, M.S.; Roh-Johnson, M.; Goodman, J.R.; Olson, B.; et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 2018, 4, eaat7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, B.; Zänker, K.S.; Dittmar, T. Cell fusion is a potent inducer of aneuploidy and drug resistance in tumor cell/ normal cell hybrids. Crit. Rev. Oncog. 2013, 18, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Carloni, V.; Mazzocca, A.; Mello, T.; Galli, A.; Capaccioli, S. Cell fusion promotes chemoresistance in metastatic colon carcinoma. Oncogene 2013, 32, 2649–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, F.; Li, C.; Wang, X.; Du, J.; Liu, K.; Li, B.; Yao, P.; Li, Y.; Zhang, S. Syncytin 1, CD9, and CD47 regulating cell fusion to form PGCCs associated with cAMP/PKA and JNK signaling pathway. Cancer Med. 2019, 8, 3047–3058. [Google Scholar] [CrossRef]
- Dittmar, T.; Schwitalla, S.; Seidel, J.; Haverkampf, S.; Reith, G.; Meyer-Staeckling, S.; Brandt, B.H.; Niggemann, B.; Zanker, K.S. Characterization of hybrid cells derived from spontaneous fusion events between breast epithelial cells exhibiting stem-like characteristics and breast cancer cells. Clin. Exp. Metastasis 2011, 28, 75–90. [Google Scholar] [CrossRef]
- Nagler, C.; Hardt, C.; Zänker, K.S.; Dittmar, T. Cocultivation of murine BMDCs with 67NR mouse mammary carcinoma cells give rise to highly drug resistant hybrid cells. Cancer Cell Int. 2011, 11, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Chen, S.; Li, C.; Ng, K.T.; Kong, C.W.; Cheng, J.; Cheng, S.H.; Li, R.A.; Lo, C.M.; Man, K.; et al. Fusion with stem cell makes the hepatocellular carcinoma cells similar to liver tumor-initiating cells. BMC Cancer 2016, 16, 56. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Zhu, Y.; Sun, Z.; Ji, R.; Zhang, X.; Xu, W.; Yuan, X.; Zhang, B.; Yan, Y.; Yin, L.; et al. Tumorigenic hybrids between mesenchymal stem cells and gastric cancer cells enhanced cancer proliferation, migration and stemness. BMC Cancer 2015, 15, 793. [Google Scholar] [CrossRef] [Green Version]
- Rappa, G.; Mercapide, J.; Lorico, A. Spontaneous formation of tumorigenic hybrids between breast cancer and multipotent stromal cells is a source of tumor heterogeneity. Am. J. Pathol. 2012, 180, 2504–2515. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Sun, X.; Wang, C.Y.; Hu, P.; Chu, C.Y.; Liu, S.; Zhau, H.E.; Chung, L.W. Spontaneous cancer-stromal cell fusion as a mechanism of prostate cancer androgen-independent progression. PLoS ONE 2012, 7, e42653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Searles, S.C.; Santosa, E.K.; Bui, J.D. Cell-cell fusion as a mechanism of DNA exchange in cancer. Oncotarget 2017, 9, 6156–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classifification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M. Vital dyes and virtual deaths. Cell Death Differ. 2013, 20, 963. [Google Scholar] [CrossRef] [Green Version]
- Eastman, A. Improving anticancer drug development begins with cell culture: Misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 2017, 8, 8854–8866. [Google Scholar] [CrossRef] [Green Version]
- Mirzayans, R.; Waters, R. DNA damage and its repair in human normal or xeroderma pigmentosum fibroblasts treated with 4-nitroquinoline 1-oxide or its 3-methyl derivative. Carcinogenesis 1982, 2, 1359–1362. [Google Scholar] [CrossRef]
- Barley, R.D.C.; Enns, L.; Paterson, M.C.; Mirzayans, R. Aberrant p21WAF1-dependent growth arrest as the possible mechanism of abnormal resistance to ultraviolet light cytotoxicity in Li-Fraumeni syndrome fifibroblast strains heterozygous for TP53 mutations. Oncogene 1998, 17, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Enns, L.; Murray, D.; Mirzayans, R. Effects of the protein kinase inhibitors wortmannin and KN62 on cellular radiosensitivity and radiation-activated S phase and G1/S checkpoints in normal human fibroblasts. Br. J. Cancer 1999, 81, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Nagarajan, A.; Uchil, P.D. Analysis of cell viability by the MTT assay. Cold Spring Harb. Protoc. 2018. [Google Scholar] [CrossRef]
- Feoktistova, M.; Geserick, P.; Leverkus, L. Crystal Violet Assay for Determining Viability of Cultured Cells. Cold Spring Harb. Protoc. 2016. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Assay | Proliferative Status | Responses Contributing to IC50 Values |
|---|---|---|
| Tetrazolium-Based Resazurin-Based (e.g., MTT, MTS, XTT, WST-1, WST-8, CellTitre-Glo) | Proliferating cultures (≥24 h treatment) |
|
| Non-proliferating cultures |
| |
| Crystal Violet Staining | Proliferating cultures (≥24 h treatment) |
|
| Non-proliferating cultures |
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirzayans, R.; Murray, D. Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. Int. J. Mol. Sci. 2020, 21, 1308. https://doi.org/10.3390/ijms21041308
Mirzayans R, Murray D. Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. International Journal of Molecular Sciences. 2020; 21(4):1308. https://doi.org/10.3390/ijms21041308
Chicago/Turabian StyleMirzayans, Razmik, and David Murray. 2020. "Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence" International Journal of Molecular Sciences 21, no. 4: 1308. https://doi.org/10.3390/ijms21041308