Oxidative Stress and Mitochondrial Abnormalities Contribute to Decreased Endothelial Nitric Oxide Synthase Expression and Renal Disease Progression in Early Experimental Polycystic Kidney Disease

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Animal Clinical and Laboratory Parameters
2.2. Renal Oxidative Stress Worsens with Disease Progression in PKD
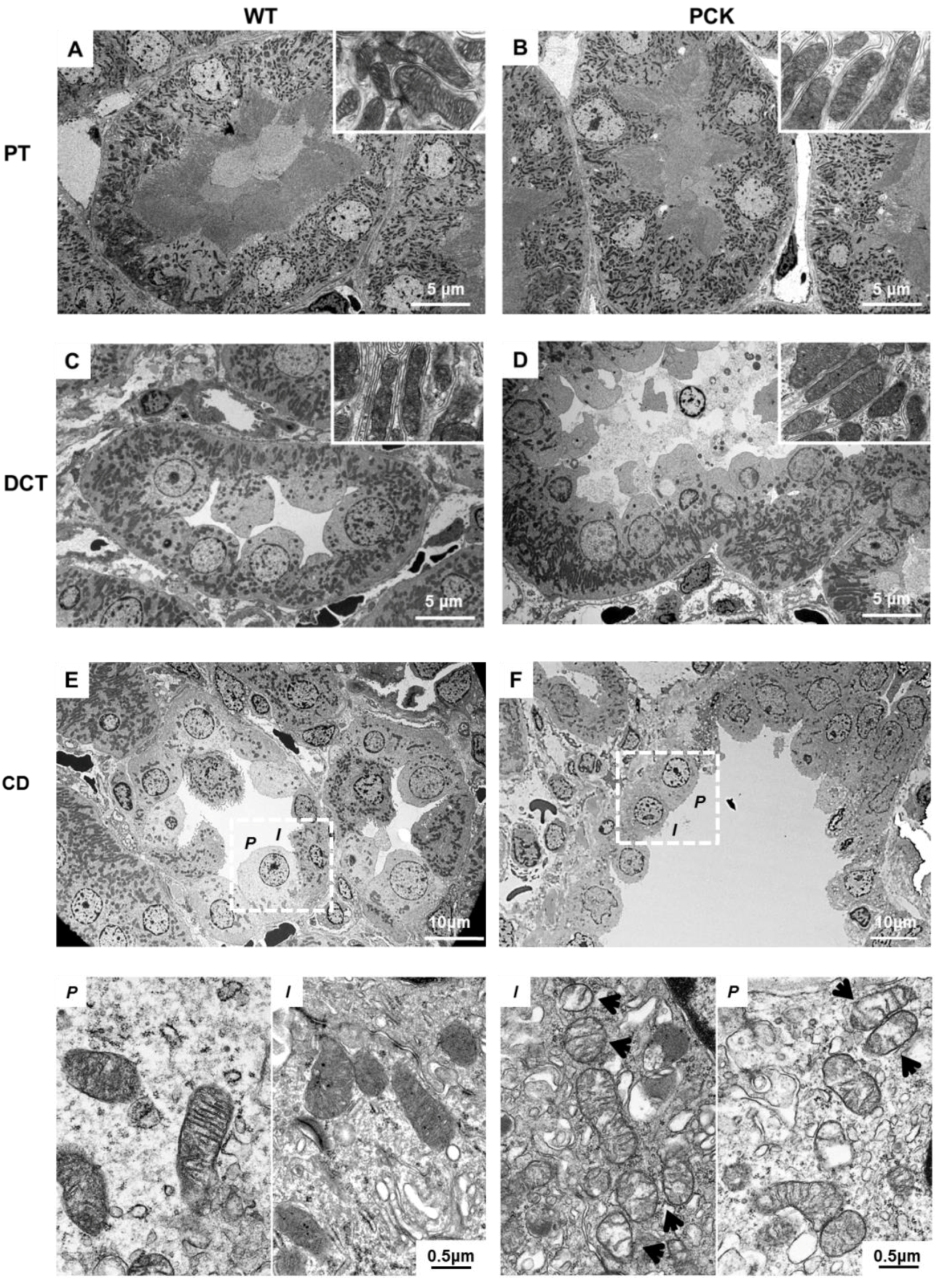
2.3. PKD is Associated with Mitochondrial Structural Abnormalities in Tubular and Endothelial Cells
2.4. Renal NOX4 is Upregulated in Tubular Epithelial and Endothelial Cells in PKD
2.5. PKD is Associated with Tubular Epithelial Cell Mitochondria Dysfunction
2.6. PKD is Associated with Intrarenal Endothelial Dysfunction and Capillary Loss
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Tissue and Blood Harvest and Analysis
4.3. Histomorphometric Analysis
4.4. Capillary Index Quantification
4.5. TEC and EC Mitochondrial Morphology and Function
4.6. Immunohistology
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADPKD | Autosomal Dominant Polycystic Kidney Disease |
| PKD | Polycystic Kidney Disease |
| ROS | Reactive Oxygen Species |
| ESRD | End-Stage Kidney Disease |
| HTN | Hypertension |
| GFR | Glomerular Filtration Rate |
| RBF | Renal blood flow |
| NO | Nitric Oxide |
| NOX4 | NAD(P)H)-oxidase complex-4 |
| WT | Wild-Type (Sprague-Dawley) |
References
- Helal, I.; Reed, B.; Mettler, P.; Mc Fann, K.; Tkachenko, O.; Yan, X.-D.; Schrier, R.W. Prevalence of cardiovascular events in patients with autosomal dominant polycystic kidney disease. Am. J. Nephrol. 2012, 36, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Helal, I.; Al-Rowaie, F.; Abderrahim, E.; Kheder, A. Update on pathogenesis, management, and treatment of hypertension in autosomal dominant polycystic kidney disease. Saudi J. kidney Dis. Transplant. 2017, 28, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W. Blood pressure in early autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2015, 372, 976–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spithoven, E.M.; Kramer, A.; Meijer, E.; Orskov, B.; Wanner, C.; Caskey, F.; Collart, F.; Finne, P.; Fogarty, D.; Groothoff, J.W.; et al. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. 2014, 86, 1244–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orskov, B.; Sorensen, V.R.; Feldt-Rasmussen, B.; Strandgaard, S. Changes in causes of death and risk of cancer in Danish patients with autosomal dominant polycystic kidney disease and end-stage renal disease. Nephrol. Dial. Transplant. 2011, 27, 1607–1613. [Google Scholar] [CrossRef] [Green Version]
- Patch, C.; Charlton, J.; Roderick, P.J.; Gulliford, M.C. Use of Antihypertensive Medications and Mortality of Patients With Autosomal Dominant Polycystic Kidney Disease: A Population-Based Study. Am. J. Kidney Dis. 2011, 57, 856–862. [Google Scholar] [CrossRef]
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368. [Google Scholar] [CrossRef]
- Shechter, M.; Matetzky, S.; Prasad, M.; Goitein, O.; Goldkorn, R.; Naroditsky, M.; Koren-Morag, N.; O Lerman, L. Endothelial function predicts 1-year adverse clinical outcome in patients hospitalized in the emergency department chest pain unit. Int. J. Cardiol. 2017, 240, 14–19. [Google Scholar] [CrossRef]
- Klawitter, J.; Reed-Gitomer, B.Y.; McFann, K.; Pennington, A.; Klawitter, J.; Abebe, K.; Klepacki, J.; Cadnapaphornchai, M.A.; Brosnahan, G.; Chonchol, M.; et al. Endothelial dysfunction and oxidative stress in polycystic kidney disease. Am. J. Physiol. Physiol. 2014, 307, F1198–F1206. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the Major Catalytic Component of an Endothelial NAD(P)H Oxidase. Circ. 2004, 109, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Boil. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowley, A.W., Jr.; Yang, C.; Zheleznova, N.N.; Staruschenko, A.; Kurth, T.; Rein, L.; Kumar, V.; Sadovnikov, K.; Dayton, A.; Hoffman, M.; et al. Evidence of the Importance of Nox4 in Production of Hypertension in Dahl Salt-Sensitive Rats. Hypertension 2016, 67, 440–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gremmels, H.; Bevers, L.M.; Fledderus, J.; Braam, B.; Van Zonneveld, A.J.; Verhaar, M.C.; Joles, J.A. Oleic acid increases mitochondrial reactive oxygen species production and decreases endothelial nitric oxide synthase activity in cultured endothelial cells. Eur. J. Pharmacol. 2015, 751, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, N.; Runge, M.S. Mitochondrial Dysfunction in Atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimoto, Y.; Inagi, R.; Yoshihara, D.; Kugita, M.; Nagao, S.; Shimizu, A.; Takeda, N.; Wake, M.; Honda, K.; Zhou, J.; et al. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Mol. Cell. Boil. 2017, 37, e00337-17. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-C.; Kurashige, M.; Liu, Y.; Terabayashi, T.; Ishimoto, Y.; Wang, T.; Choudhary, V.; Hobbs, R.; Liu, L.-K.; Lee, P.-H.; et al. A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci. Rep. 2018, 8, 2743. [Google Scholar] [CrossRef]
- Kuo, I.Y.; Brill, A.L.; Lemos, F.O.; Jiang, J.Y.; Falcone, J.L.; Kimmerling, E.P.; Cai, Y.; Dong, K.; Kaplan, D.L.; Wallace, D.P.; et al. Polycystin 2 regulates mitochondrial Ca2+ signaling, bioenergetics, and dynamics through mitofusin 2. Sci. Signal. 2019, 12, eaat7397. [Google Scholar] [CrossRef]
- Lager, N.J.; Qian, Q.; Bengal, R.J.; Ishibashi, M.; Torres, V.E. The pck rat: A new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int. 2001, 59, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gattone, V.; Harris, P.C.; Torres, V.E. Effectiveness of Vasopressin V2 Receptor Antagonists OPC-31260 and OPC-41061 on Polycystic Kidney Disease Development in the PCK Rat. J. Am. Soc. Nephrol. 2005, 16, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wu, Y.; Ward, C.J.; Harris, P.C.; Torres, V.E. Vasopressin directly regulates cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 19, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ye, H.; Ward, C.J.; Chu, J.Y.S.; Masyuk, T.V.; LaRusso, N.F.; Harris, P.C.; Chow, B.K.C.; Torres, V.E. Insignificant effect of secretin in rodent models of polycystic kidney and liver disease. Am. J. Physiol. Physiol. 2012, 303, F1089–F1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopp, K.; Wang, X.; Ye, H.; Irazabal, M.V.; Harris, P.C.; Torres, V.E. Effects of hydration in rats and mice with polycystic kidney disease. Am. J. Physiol. Physiol. 2014, 308, F261–F266. [Google Scholar] [CrossRef] [PubMed]
- Iannaccone, P.M.; Jacob, H.J. Rats! Dis. Models Mech. 2009, 2, 206–210. [Google Scholar] [CrossRef] [Green Version]
- Bracken, C.; Beauverger, P.; Duclos, O.; Russo, R.J.; Rogers, K.A.; Husson, H.; Natoli, T.A.; Ledbetter, S.R.; Janiak, P.; Ibraghimov-Beskrovnaya, O.; et al. CaMKII as a pathological mediator of ER stress, oxidative stress, and mitochondrial dysfunction in a murine model of nephronophthisis. Am. J. Physiol. Physiol. 2016, 310, F1414–F1422. [Google Scholar] [CrossRef] [Green Version]
- Serrander, L.; Cartier, L.; Bedard, K.; Banfi, B.; Lardy, B.; Plastre, O.; Sienkiewicz, A.; Forró, L.; Schlegel, W.; Krause, K.-H. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007, 406, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Block, K.; Gorin, Y.; Abboud, H. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. USA 2009, 106, 14385–14390. [Google Scholar] [CrossRef] [Green Version]
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hébert, R.L. NADPH Oxidases, Reactive Oxygen Species, and the Kidney: Friend and Foe. J. Am. Soc. Nephrol. 2013, 24, 1512–1518. [Google Scholar] [CrossRef]
- Jha, J.C.; Gray, S.P.; Barit, D.; Okabe, J.; El-Osta, A.; Namikoshi, T.; Thallas-Bonke, V.; Wingler, K.; Szyndralewiez, C.; Heitz, F.; et al. Genetic Targeting or Pharmacologic Inhibition of NADPH Oxidase Nox4 Provides Renoprotection in Long-Term Diabetic Nephropathy. J. Am. Soc. Nephrol. 2014, 25, 1237–1254. [Google Scholar] [CrossRef] [Green Version]
- Thallas-Bonke, V.; Jandeleit-Dahm, K.A.; Cooper, M.E. Nox-4 and progressive kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 74–80. [Google Scholar] [CrossRef]
- You, Y.-H.; Quach, T.; Saito, R.; Pham, J.; Sharma, K. Metabolomics Reveals a Key Role for Fumarate in Mediating the Effects of NADPH Oxidase 4 in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2015, 27, 466–481. [Google Scholar] [CrossRef] [PubMed]
- Vendrov, A.E.; Vendrov, K.C.; Smith, A.; Yuan, J.; Sumida, A.; Robidoux, J.; Runge, M.S.; Madamanchi, N.R. NOX4 NADPH Oxidase-Dependent Mitochondrial Oxidative Stress in Aging-Associated Cardiovascular Disease. Antioxidants Redox Signal. 2015, 23, 1389–1409. [Google Scholar] [CrossRef] [PubMed]
- Bernard, K.; Logsdon, N.J.; Miguel, V.; Benavides, G.A.; Zhang, J.; Carter, A.B.; Darley-Usmar, V.; Thannickal, V.J. NADPH Oxidase 4 (Nox4) Suppresses Mitochondrial Biogenesis and Bioenergetics in Lung Fibroblasts via a Nuclear Factor Erythroid-derived 2-like 2 (Nrf2)-dependent Pathway. J. Boil. Chem. 2017, 292, 3029–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Boil. 2014, 2, 702–714. [Google Scholar] [CrossRef] [Green Version]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Padovano, V.; Kuo, I.Y.; Stavola, L.K.; Aerni, H.R.; Flaherty, B.J.; Chapin, H.C.; Ma, M.; Somlo, S.; Boletta, A.; Ehrlich, B.E.; et al. The polycystins are modulated by cellular oxygen-sensing pathways and regulate mitochondrial function. Mol. Boil. Cell 2017, 28, 261–269. [Google Scholar] [CrossRef]
- Hajarnis, S.; Lakhia, R.; Yheskel, M.; Williams, D.; Sorourian, M.; Liu, X.; Aboudehen, K.; Zhang, S.; Kersjes, K.; Galasso, R.; et al. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat. Commun. 2017, 8, 14395. [Google Scholar] [CrossRef]
- Podrini, C.; Rowe, I.; Pagliarini, R.; Costa, A.; Chiaravalli, M.; Di Meo, I.; Kim, H.; Distefano, G.; Tiranti, V.; Qian, F.; et al. Dissection of metabolic reprogramming in polycystic kidney disease reveals coordinated rewiring of bioenergetic pathways. Commun. Boil. 2018, 1, 194. [Google Scholar] [CrossRef] [Green Version]
- Kluge, M.A.; Fetterman, J.L.; Vita, J.A. Mitochondria and endothelial function. Circ. Res. 2013, 112, 1171–1188. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Lanza, I.R.; Nair, K.S. Chapter 20: Functional Assessment of Isolated Mitochondria in Vitro. Methods Enzym. 2009, 457, 349–372. [Google Scholar]
- Lanza, I.R.; Nair, K.S. Mitochondrial metabolic function assessed in vivo and in vitro. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eirin, A.; Li, Z.; Zhang, X.; Krier, J.D.; Woollard, J.R.; Zhu, X.-Y.; Tang, H.; Herrmann, S.M.; Lerman, A.; Textor, S.C.; et al. A Mitochondrial Permeability Transition Pore Inhibitor Improves Renal Outcomes After Revascularization in Experimental Atherosclerotic Renal Artery Stenosis. Hypertension 2012, 60, 1242–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Characteristic | 4 Week | 12 Week | ||
|---|---|---|---|---|
| WT | PCK | WT | PCK | |
| Number of animals | 16 | 16 | 16 | 16 |
| Body Weight (g) | 152.0 ± 8.4 | 149.7 ± 7.2 | 379.9 ± 105.6 † | 381.8 ± 91.4 † |
| KW/BW (%) | 1.09 ± 0.08 | 1.48 ± 0.09 **** | 0.79 ± 0.11 † | 1.84 ± 0.22 **** † |
| Kidney Cystic Index (%) | 0.05 ± 0.03 | 15.54 ± 2.92 **** | 0.15 ± 0.11 | 21.4 ± 3.56 **** † |
| Fibrotic Index (%) | 0.40 ± 0.03 | 0.42 ± 0.04 | 0.35 ± 0.21 | 3.51 ± 1.76 **** † |
| Urine output (24 h, mL) | 5.1 ± 0.5 | 4.8 ± 0.7 | 12.8 ± 3.3 † | 13.2 ± 4.3 † |
| BUN (mg/dL) | 12.3 ± 2.2 | 12.6 ± 1.9 | 15.9 ± 1.8 ‡ | 20.1 ± 3.7 **** † |
| Plasma creatinine (mg/dL) | 0.36 ± 0.05 | 0.35 ± 0.10 | 0.36 ± 0.10 | 0.38 ± 0.13 |
| Blood Pressure (mmHg) | 109.4 ± 2.3 | 110.3 ± 2.1 | 116.2 ± 3.1 † | 118.3 ± 2.9 † |
| Animal Characteristic | 4 Week | 12 Week | ||
|---|---|---|---|---|
| WT | PCK | WT | PCK | |
| Number of animals | 16 | 16 | 16 | 16 |
| Capillary Index (Cx) | 10.9 ± 0.8 | 10.5 ± 0.6 | 11.3 ± 0.6 | 9.3 ± 0.8 **** ‡ |
| Capillary Index (Med) | 9.4 ± 0.4 | 9.2 ± 0.5 | 10.0 ± 0.5 + | 8.3 ± 0.5 **** † |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kahveci, A.S.; Barnatan, T.T.; Kahveci, A.; Adrian, A.E.; Arroyo, J.; Eirin, A.; Harris, P.C.; Lerman, A.; Lerman, L.O.; Torres, V.E.; et al. Oxidative Stress and Mitochondrial Abnormalities Contribute to Decreased Endothelial Nitric Oxide Synthase Expression and Renal Disease Progression in Early Experimental Polycystic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 1994. https://doi.org/10.3390/ijms21061994
Kahveci AS, Barnatan TT, Kahveci A, Adrian AE, Arroyo J, Eirin A, Harris PC, Lerman A, Lerman LO, Torres VE, et al. Oxidative Stress and Mitochondrial Abnormalities Contribute to Decreased Endothelial Nitric Oxide Synthase Expression and Renal Disease Progression in Early Experimental Polycystic Kidney Disease. International Journal of Molecular Sciences. 2020; 21(6):1994. https://doi.org/10.3390/ijms21061994
Chicago/Turabian StyleKahveci, Alp S., Tania T. Barnatan, Ali Kahveci, Alexis E. Adrian, Jennifer Arroyo, Alfonso Eirin, Peter C. Harris, Amir Lerman, Lilach O. Lerman, Vicente E. Torres, and et al. 2020. "Oxidative Stress and Mitochondrial Abnormalities Contribute to Decreased Endothelial Nitric Oxide Synthase Expression and Renal Disease Progression in Early Experimental Polycystic Kidney Disease" International Journal of Molecular Sciences 21, no. 6: 1994. https://doi.org/10.3390/ijms21061994