Calcineurin Controls Expression of EAAT1/GLAST in Mouse and Human Cultured Astrocytes through Dynamic Regulation of Protein Synthesis and Degradation


 ,
,  and
and 
Abstract
1. Introduction
2. Results
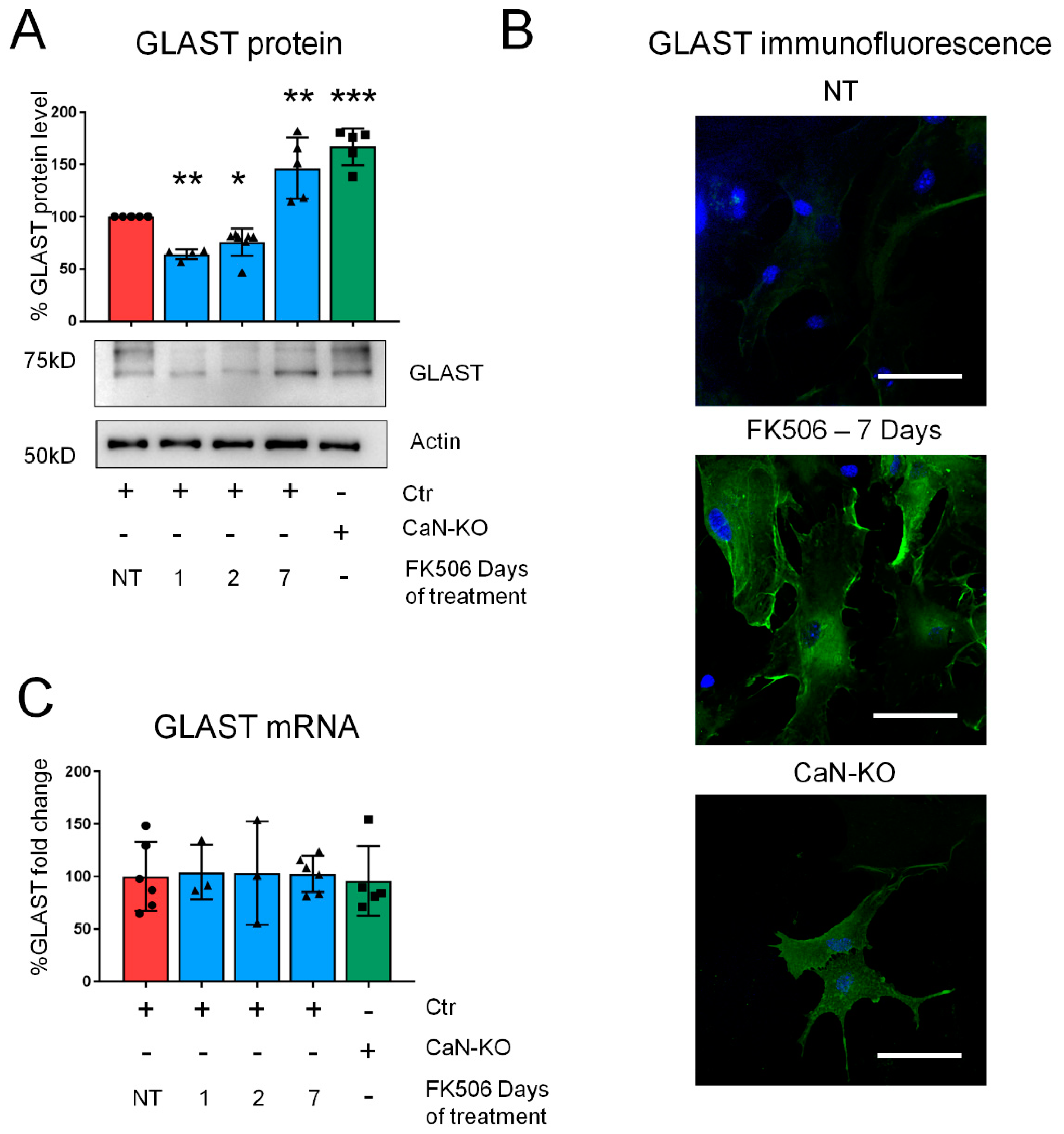
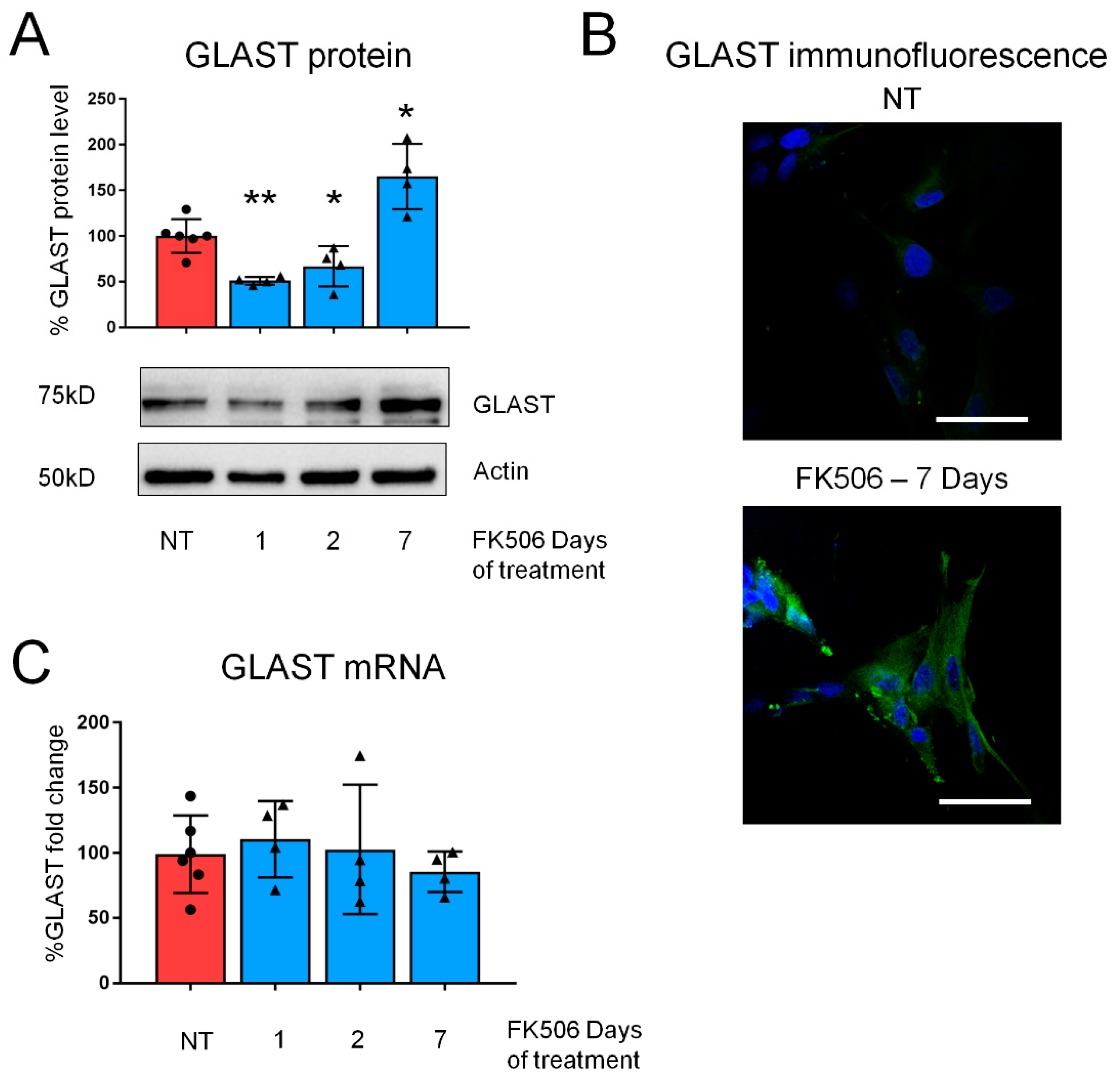
2.1. Pharmacological CaN Inhibition Results in a Dynamic Modulation of GLAST Protein in Both Mouse and Human Astrocytes
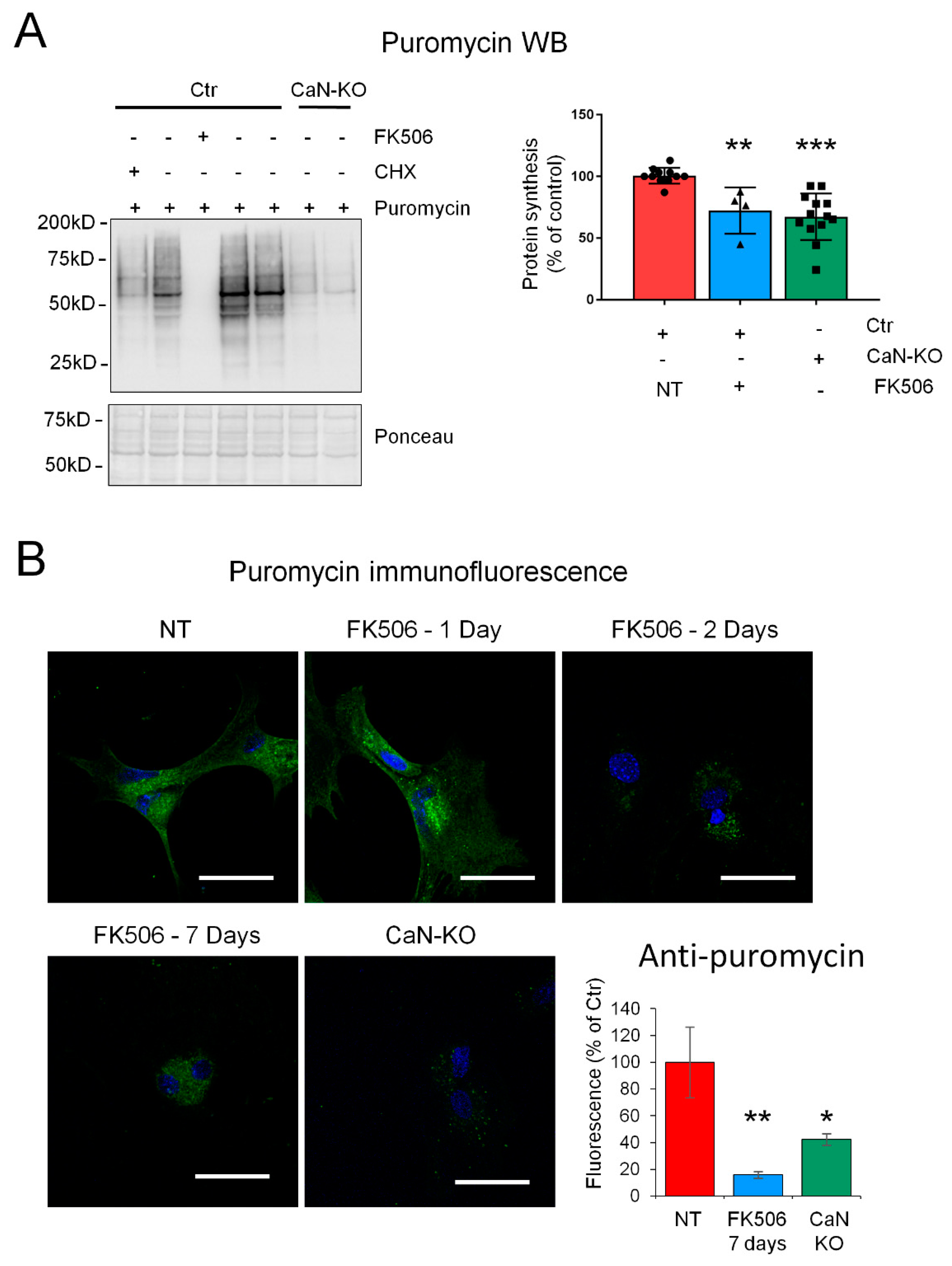
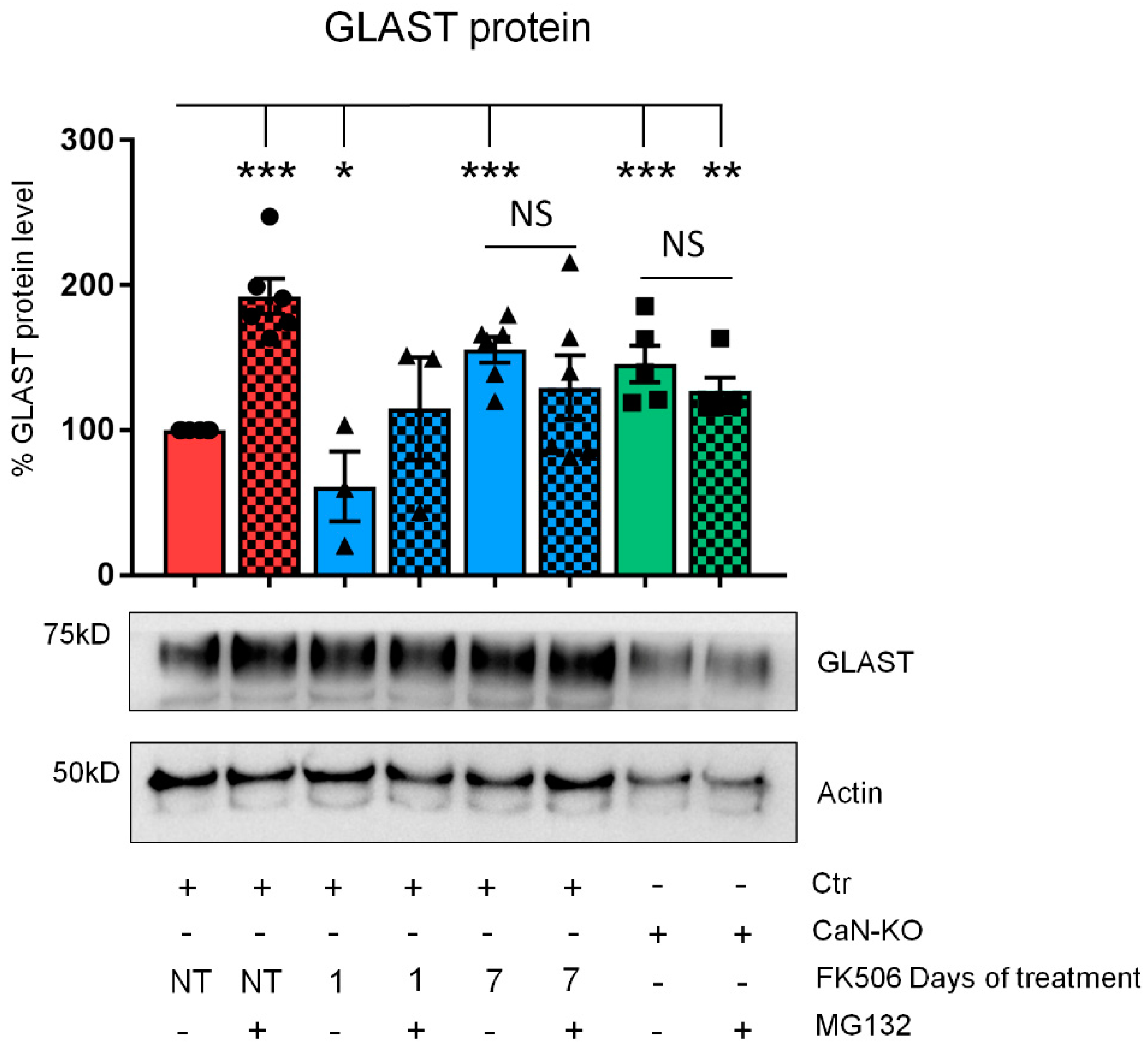
2.2. CaN Modulates GLAST Protein Expression through the Regulation of the Equilibrium between Protein Synthesis and Degradation
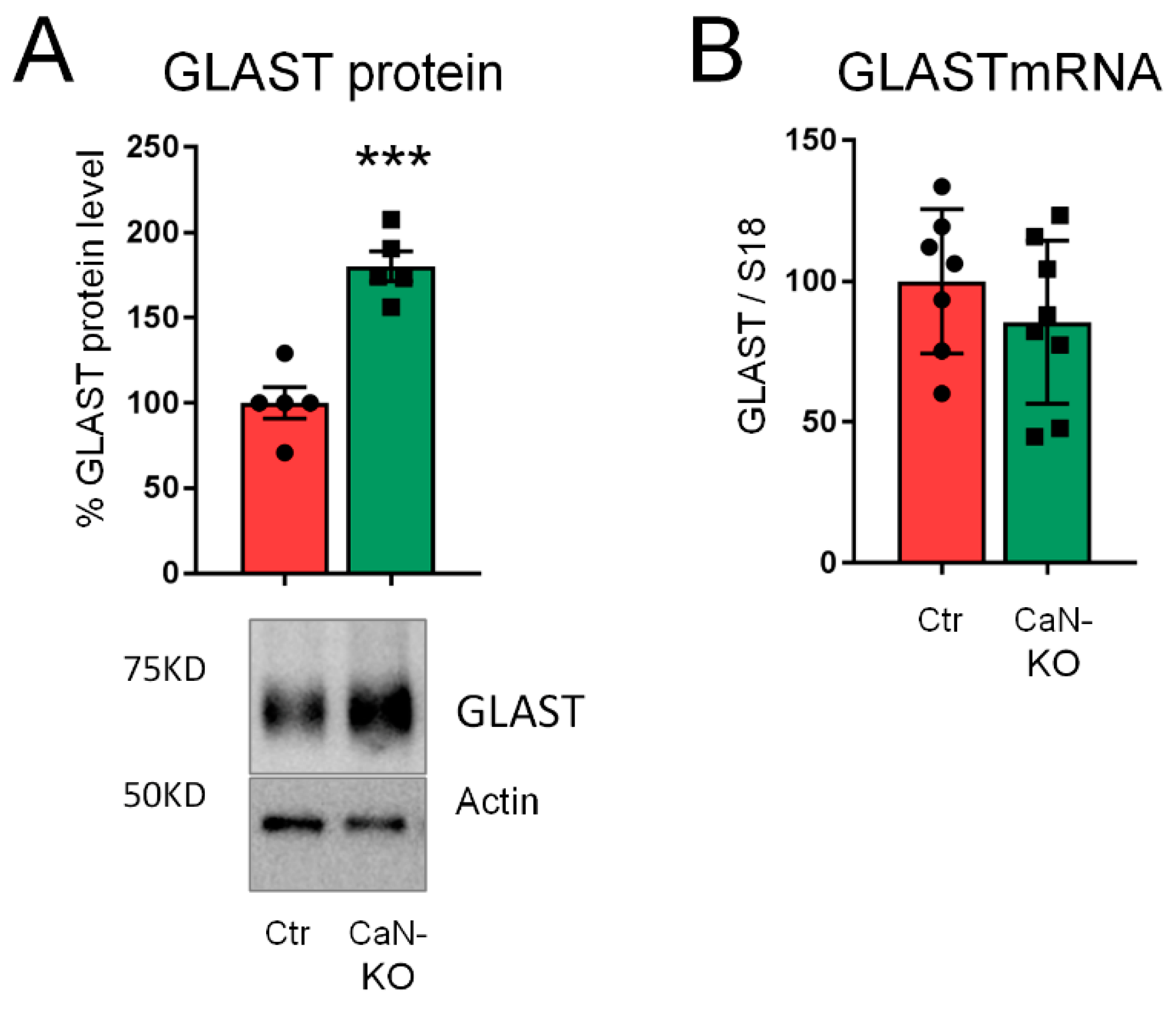
2.3. Upregulation of GLAST Protein in Astroglial CaN-KO Hippocampal Synaptosomes
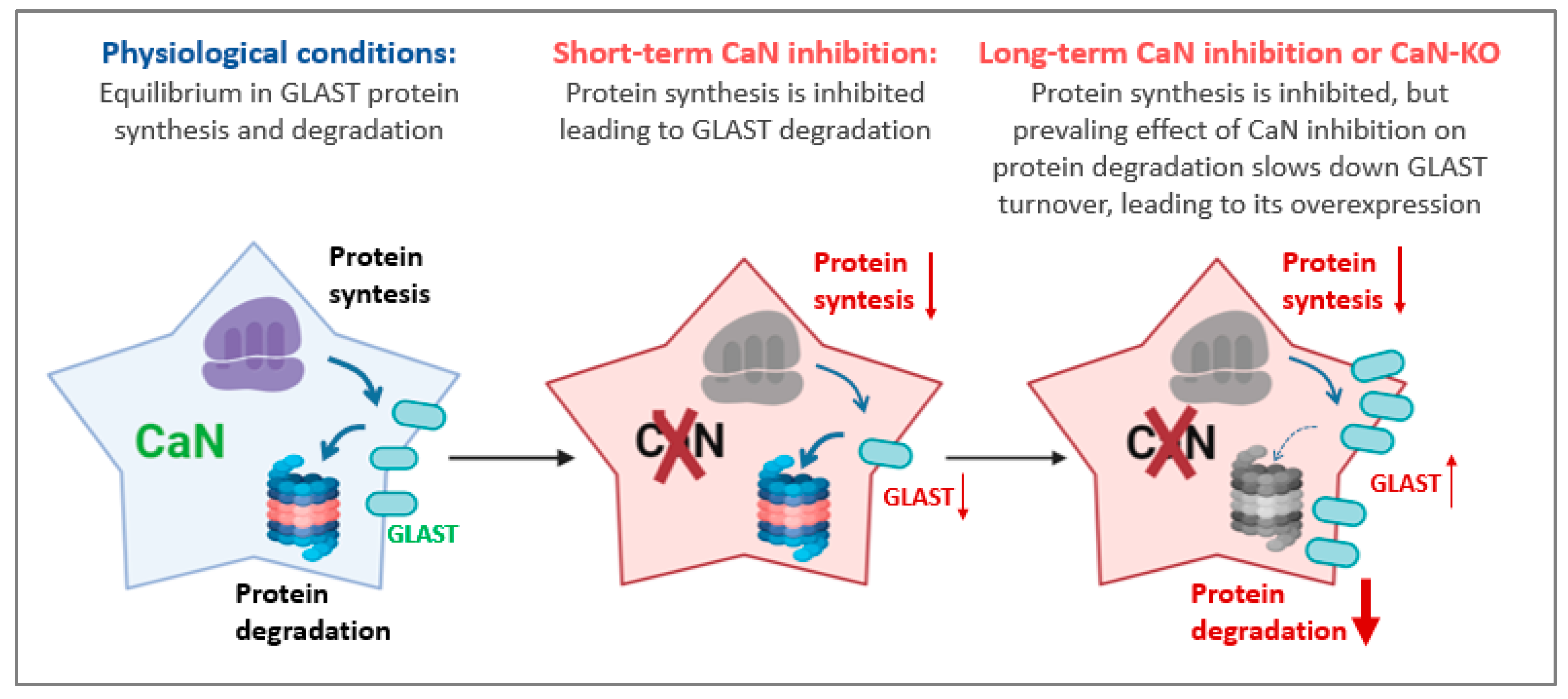
3. Discussion
4. Materials and Methods
4.1. Astrocyte-Specific CaN KO Mice
4.2. Mouse Astrocytic Primary Cultures
4.3. Fetal Human Primary Astrocytes
4.4. Pharmacological Treatments
4.5. Puromycin Incorporation Method (Surface Sensing of Translation, SUnSET)
4.6. Immunofluorescence and Confocal Microscopy
4.7. Preparation of Synaptosomes
4.8. Cell lysis and Western blot
4.9. RNA Extraction and Real-Time PCR
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Nicholls, D.; Attwell, D. The release and uptake of excitatory amino acids. Trends Pharmacol. Sci. 1990, 11, 462–468. [Google Scholar] [CrossRef]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 5671. [Google Scholar] [CrossRef]
- Garlin, A.B.; Sinor, A.D.; Sinor, J.D.; Jee, S.H.; Grinspan, J.B.; Robinson, M.B. Pharmacology of sodium-dependent high-affinity L-[3H]glutamate transport in glial cultures. J. Neurochem. 1995, 64, 2572–2580. [Google Scholar] [CrossRef]
- Swanson, R.A.; Liu, J.; Miller, J.W.; Rothstein, J.D.; Farrell, K.; Stein, B.A.; Longuemare, M.C. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 932–940. [Google Scholar] [CrossRef]
- Tapella, L.; Soda, T.; Mapelli, L.; Bortolotto, V.; Bondi, H.; Ruffinatti, F.A.; Dematteis, G.; Stevano, A.; Dionisi, M.; Ummarino, S.; et al. Deletion of calcineurin from GFAP-expressing astrocytes impairs excitability of cerebellar and hippocampal neurons through astroglial Na+ /K+ ATPase. Glia 2020, 68, 543–560. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]
- Bristot Silvestrin, R.; Bambini-Junior, V.; Galland, F.; Daniele Bobermim, L.; Quincozes-Santos, A.; Torres Abib, R.; Zanotto, C.; Batassini, C.; Brolese, G.; Gonçalves, C.-A.; et al. Animal model of autism induced by prenatal exposure to valproate: altered glutamate metabolism in the hippocampus. Brain Res. 2013, 1495, 52–60. [Google Scholar] [CrossRef]
- Martinez-Lozada, Z.; Guillem, A.M.; Robinson, M.B. Transcriptional Regulation of Glutamate Transporters: From Extracellular Signals to Transcription Factors. Adv. Pharmacol. San Diego Calif 2016, 76, 103–145. [Google Scholar]
- Lee, E.; Karki, P.; Johnson, J.; Hong, P.; Aschner, M. Manganese Control of Glutamate Transporters’ Gene Expression. Adv. Neurobiol. 2017, 16, 1–12. [Google Scholar]
- Abdul, H.M.; Sama, M.A.; Furman, J.L.; Mathis, D.M.; Beckett, T.L.; Weidner, A.M.; Patel, E.S.; Baig, I.; Murphy, M.P.; LeVine, H.; et al. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 12957–12969. [Google Scholar] [CrossRef]
- Lim, D.; Rocchio, F.; Lisa, M.; Fcancesco, M. From Pathology to Physiology of Calcineurin Signalling in Astrocytes. Opera Med. Physiol. 2016, 2, 43–61. [Google Scholar]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 1997, 15, 707–747. [Google Scholar] [CrossRef]
- Lee, J.I.; Burckart, G.J. Nuclear factor kappa B: important transcription factor and therapeutic target. J. Clin. Pharmacol. 1998, 38, 981–993. [Google Scholar] [CrossRef]
- Li, H.; Rao, A.; Hogan, P.G. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol. 2011, 21, 91–103. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Chaudhry, F.A.; Lehre, K.P.; van Lookeren Campagne, M.; Ottersen, O.P.; Danbolt, N.C.; Storm-Mathisen, J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 1995, 15, 711–720. [Google Scholar] [CrossRef]
- Rudy, C.C.; Hunsberger, H.C.; Weitzner, D.S.; Reed, M.N. The Role of the Tripartite Glutamatergic Synapse in the Pathophysiology of Alzheimer’s Disease. Aging Dis. 2015, 6, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, J.; Brito, V.; Küppers, E.; Beyer, C. Regulation of glutamate transporter GLAST and GLT-1 expression in astrocytes by estrogen. Brain Res. Mol. Brain Res. 2005, 138, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ikegaya, Y.; Matsuura, S.; Kanai, Y.; Endou, H.; Matsuki, N. Transient upregulation of the glial glutamate transporter GLAST in response to fibroblast growth factor, insulin-like growth factor and epidermal growth factor in cultured astrocytes. J. Cell Sci. 2001, 114, 3717–3725. [Google Scholar] [PubMed]
- Espinoza-Rojo, M.; López-Bayghen, E.; Ortega, A. GLAST: gene expression regulation by phorbol esters. Neuroreport 2000, 11, 2827–2832. [Google Scholar] [CrossRef]
- Wang, Z.; Li, W.; Mitchell, C.K.; Carter-Dawson, L. Activation of protein kinase C reduces GLAST in the plasma membrane of rat Müller cells in primary culture. Vis. Neurosci. 2003, 20, 611–619. [Google Scholar] [CrossRef]
- Piao, C.; Ralay Ranaivo, H.; Rusie, A.; Wadhwani, N.; Koh, S.; Wainwright, M.S. Thrombin decreases expression of the glutamate transporter GLAST and inhibits glutamate uptake in primary cortical astrocytes via the Rho kinase pathway. Exp. Neurol. 2015, 273, 288–300. [Google Scholar] [CrossRef]
- Perego, C.; Vanoni, C.; Bossi, M.; Massari, S.; Basudev, H.; Longhi, R.; Pietrini, G. The GLT-1 and GLAST glutamate transporters are expressed on morphologically distinct astrocytes and regulated by neuronal activity in primary hippocampal cocultures. J. Neurochem. 2000, 75, 1076–1084. [Google Scholar] [CrossRef]
- Cho, S.; Muthukumar, A.K.; Stork, T.; Coutinho-Budd, J.C.; Freeman, M.R. Focal adhesion molecules regulate astrocyte morphology and glutamate transporters to suppress seizure-like behavior. Proc. Natl. Acad. Sci. USA 2018, 115, 11316–11321. [Google Scholar] [CrossRef]
- Tai, Y.-H.; Tsai, R.-Y.; Wang, Y.-H.; Cherng, C.-H.; Tao, P.-L.; Liu, T.-M.; Wong, C.-S. Amitriptyline induces nuclear transcription factor-kappaB-dependent glutamate transporter upregulation in chronic morphine-infused rats. Neuroscience 2008, 153, 823–831. [Google Scholar] [CrossRef]
- Karki, P.; Kim, C.; Smith, K.; Son, D.-S.; Aschner, M.; Lee, E. Transcriptional Regulation of the Astrocytic Excitatory Amino Acid Transporter 1 (EAAT1) via NF-κB and Yin Yang 1 (YY1). J. Biol. Chem. 2015, 290, 23725–23737. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Fernandez, S.; Carrero, P.; Garcia-Garcia, M.; Torres-Aleman, I. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 8745–8756. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.M.; Jimenez, S.; Mecha, M.; Dávila, D.; Guaza, C.; Vitorica, J.; Torres-Aleman, I. Regulation of the phosphatase calcineurin by insulin-like growth factor I unveils a key role of astrocytes in Alzheimer’s pathology. Mol. Psychiatry 2012, 17, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Iyer, A.; Ronco, V.; Grolla, A.A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Furman, J.L.; Norris, C.M. Calcineurin and glial signaling: neuroinflammation and beyond. J. Neuroinflammation 2014, 11, 158. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Meyer, J.G.; Schilling, B. Protein Turnover in Aging and Longevity. Proteomics 2018, 18, e1700108. [Google Scholar] [CrossRef]
- Sans, M.D.; Williams, J.A. Calcineurin is required for translational control of protein synthesis in rat pancreatic acini. Am. J. Physiol. Cell Physiol. 2004, 287, C310–C319. [Google Scholar] [CrossRef] [PubMed]
- Aufschnaiter, A.; Kohler, V.; Büttner, S. Taking out the garbage: cathepsin D and calcineurin in neurodegeneration. Neural Regen. Res. 2017, 12, 1776–1779. [Google Scholar]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef]
- Lapierre, L.R.; Kumsta, C.; Sandri, M.; Ballabio, A.; Hansen, M. Transcriptional and epigenetic regulation of autophagy in aging. Autophagy 2015, 11, 867–880. [Google Scholar] [CrossRef]
- Mukherjee, A.; Soto, C. Role of calcineurin in neurodegeneration produced by misfolded proteins and endoplasmic reticulum stress. Curr. Opin. Cell Biol. 2011, 23, 223–230. [Google Scholar] [CrossRef]
- Shah, S.Z.A.; Hussain, T.; Zhao, D.; Yang, L. A central role for calcineurin in protein misfolding neurodegenerative diseases. Cell. Mol. Life Sci. CMLS 2017, 74, 1061–1074. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-H.; Kedar, V.; Zhang, C.; McDonough, H.; Arya, R.; Wang, D.-Z.; Patterson, C. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J. Clin. Invest. 2004, 114, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Langenbacher, A.D.; Huang, J.; Wang, K.; Otto, G.; Geisler, R.; Wang, Y.; Chen, J.-N. The Calcineurin-FoxO-MuRF1 signaling pathway regulates myofibril integrity in cardiomyocytes. eLife 2017, 6, e27955. [Google Scholar] [CrossRef] [PubMed]
- Bollo, M.; Paredes, R.M.; Holstein, D.; Zheleznova, N.; Camacho, P.; Lechleiter, J.D. Calcineurin interacts with PERK and dephosphorylates calnexin to relieve ER stress in mammals and frogs. PloS One 2010, 5, e11925. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Holstein, D.M.; Aime, S.; Bollo, M.; Lechleiter, J.D. Calcineurin β protects brain after injury by activating the unfolded protein response. Neurobiol. Dis. 2016, 94, 139–156. [Google Scholar] [CrossRef]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schätzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866. [Google Scholar] [CrossRef]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef]
- Korotkov, A.; Broekaart, D.W.M.; Banchaewa, L.; Pustjens, B.; van Scheppingen, J.; Anink, J.J.; Baayen, J.C.; Idema, S.; Gorter, J.A.; van Vliet, E.A.; et al. microRNA-132 is overexpressed in glia in temporal lobe epilepsy and reduces the expression of pro-epileptogenic factors in human cultured astrocytes. Glia 2020, 68, 60–75. [Google Scholar] [CrossRef]
- Gillardon, F. Differential mitochondrial protein expression profiling in neurodegenerative diseases. Electrophoresis 2006, 27, 2814–2818. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Name | Gene Name | Forward/Reverse | Accession No. |
|---|---|---|---|
| S18 | Rps18 | TGCGAGTACTCAACACCAACA | NM_011296 |
| mouse and human | CTGCTTTCCTCAACACCACA | NM_022551.3 | |
| EAAT1/GLAST mouse | Slc1a3 | AATGCCTTCGTTCTGCTCAC | NM_148938 |
| ATCCTCATGAGAAGCTCCCC | |||
| EAAT1/GLAST human | Slc1a3 | GTTGCTCCGTTGTACCTGCT | NM_004172.4 |
| GGAATCACCCACAGAAAGCC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dematteis, G.; Restelli, E.; Chiesa, R.; Aronica, E.; Genazzani, A.A.; Lim, D.; Tapella, L. Calcineurin Controls Expression of EAAT1/GLAST in Mouse and Human Cultured Astrocytes through Dynamic Regulation of Protein Synthesis and Degradation. Int. J. Mol. Sci. 2020, 21, 2213. https://doi.org/10.3390/ijms21062213
Dematteis G, Restelli E, Chiesa R, Aronica E, Genazzani AA, Lim D, Tapella L. Calcineurin Controls Expression of EAAT1/GLAST in Mouse and Human Cultured Astrocytes through Dynamic Regulation of Protein Synthesis and Degradation. International Journal of Molecular Sciences. 2020; 21(6):2213. https://doi.org/10.3390/ijms21062213
Chicago/Turabian StyleDematteis, Giulia, Elena Restelli, Roberto Chiesa, Eleonora Aronica, Armando A Genazzani, Dmitry Lim, and Laura Tapella. 2020. "Calcineurin Controls Expression of EAAT1/GLAST in Mouse and Human Cultured Astrocytes through Dynamic Regulation of Protein Synthesis and Degradation" International Journal of Molecular Sciences 21, no. 6: 2213. https://doi.org/10.3390/ijms21062213
APA StyleDematteis, G., Restelli, E., Chiesa, R., Aronica, E., Genazzani, A. A., Lim, D., & Tapella, L. (2020). Calcineurin Controls Expression of EAAT1/GLAST in Mouse and Human Cultured Astrocytes through Dynamic Regulation of Protein Synthesis and Degradation. International Journal of Molecular Sciences, 21(6), 2213. https://doi.org/10.3390/ijms21062213