“Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres


 , , and
, , and
Abstract
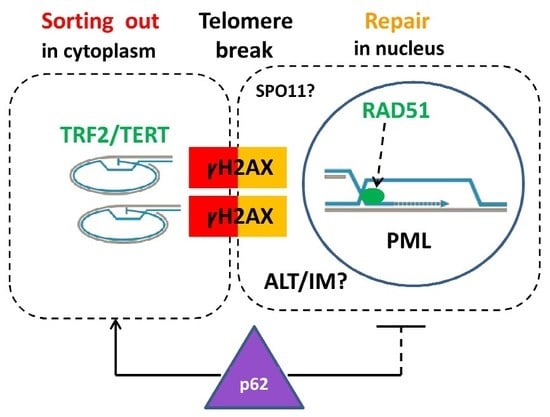
:
1. Introduction
2. Results
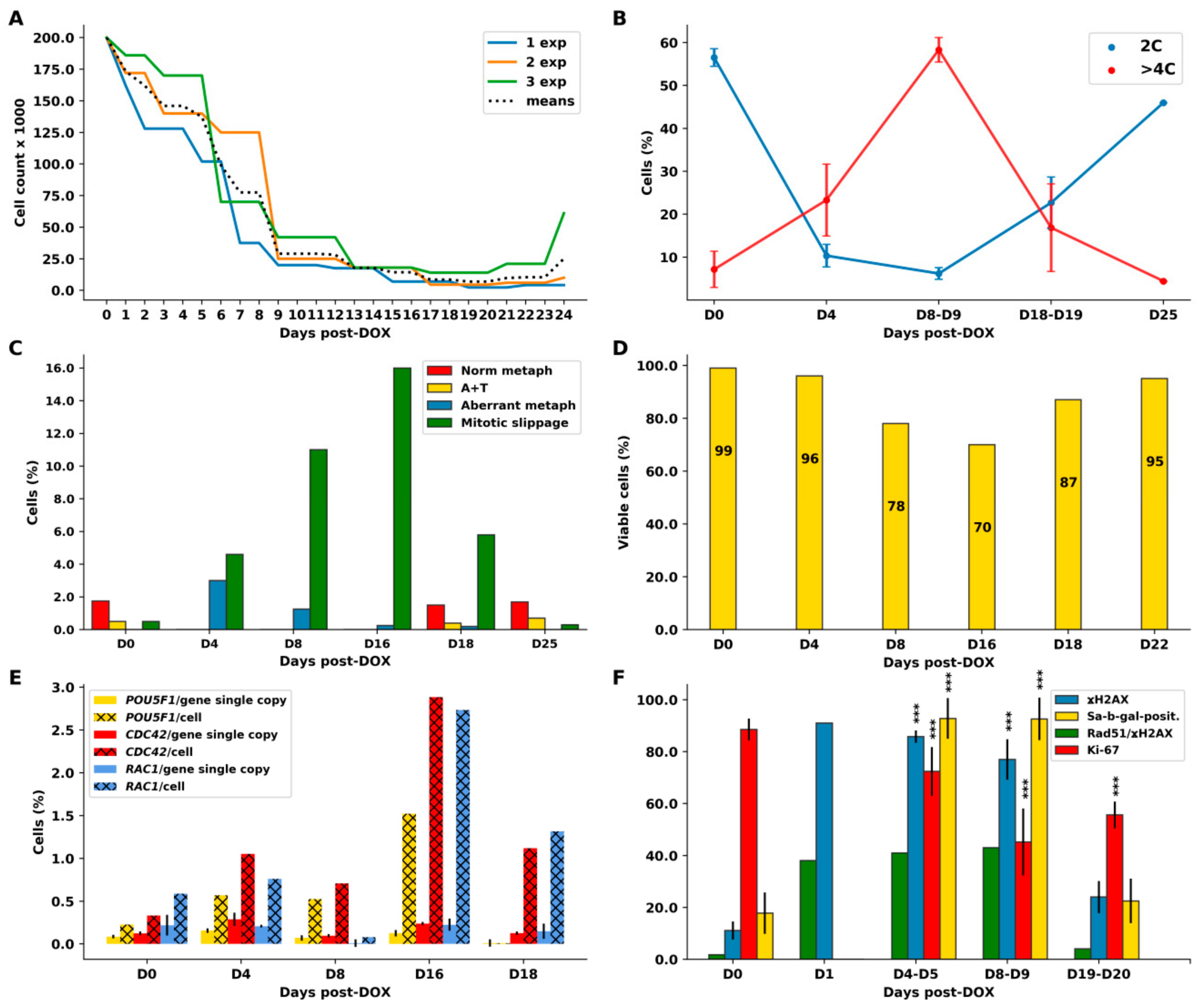
2.1. Breast Cancer MDA-MB-231 Cell Line, before and after Doxorubicin (DOX) Treatment: The Phenotypes, Cell Growth, and Outlines of the Findings
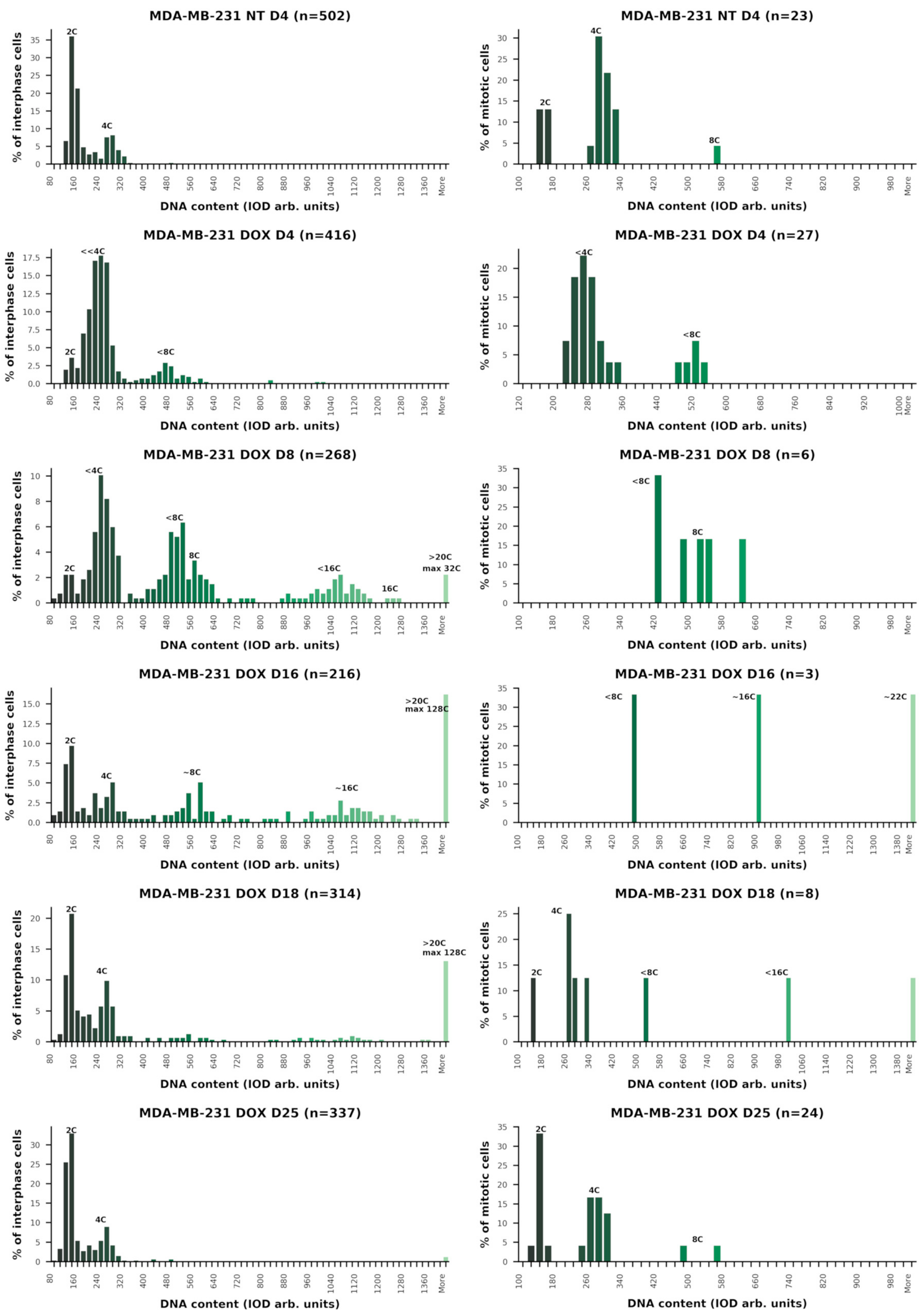
2.2. Cell Cycle Dynamics and Mitotic Slippage (MS) after DOX-Treatment
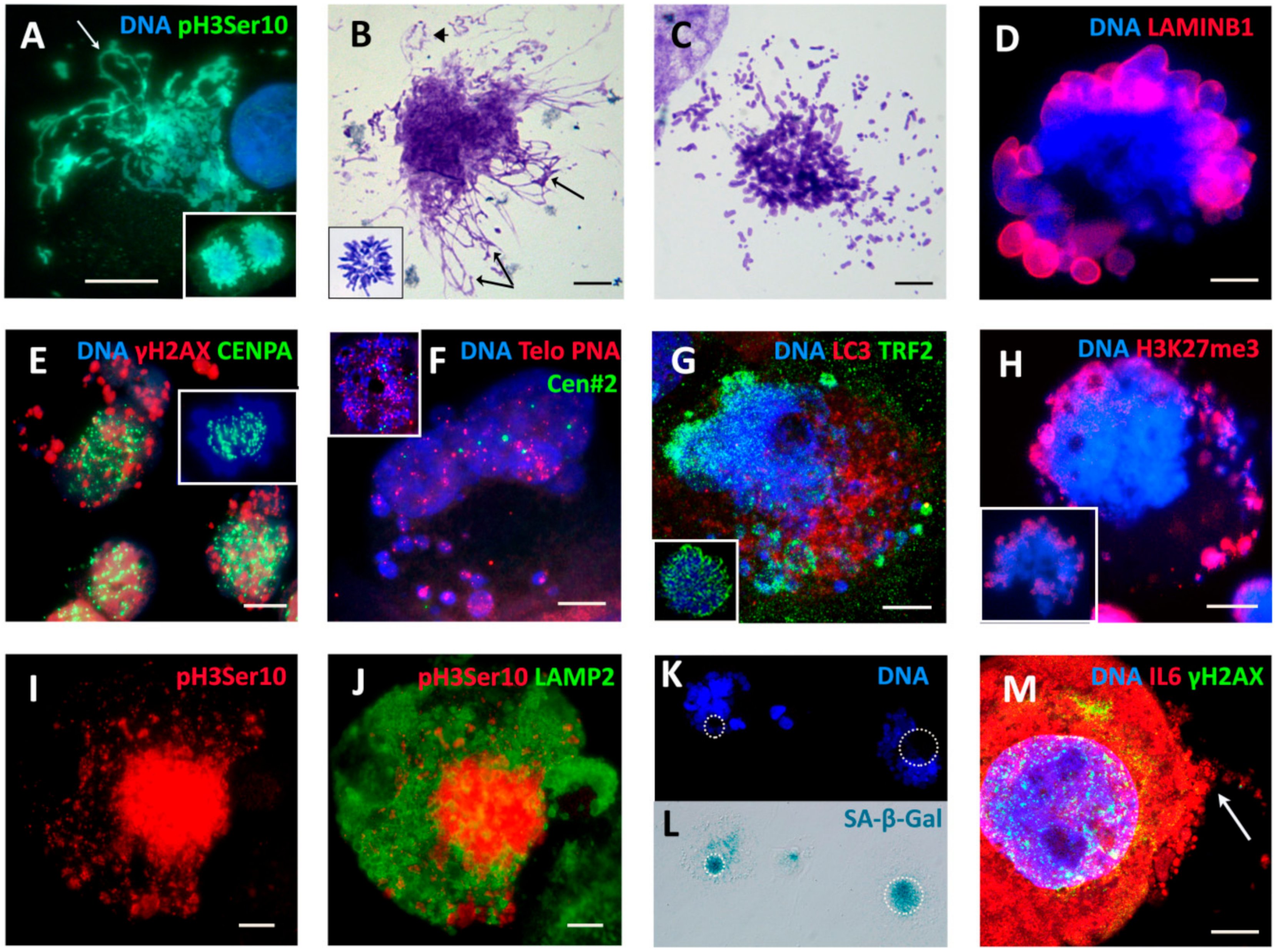
2.3. The Amoeboid Transition of Polyploidized Breast Cancer Cells and Budding of the Mitotic Progeny
2.4. Extranuclear DNA Released by MS in DOX-Treated Cells Contains Telomere Heterochromatin Enriched with the TRF2-Shelterin but Not Centromeres
2.5. The Features of Cellular Senescence in the MDA-MB-231-DOX-Treated Cells with Persistent DNA Damage, Ki-67 Positivity, and HR DNA Repair are Related to MS
2.6. Homologous Recombination (HR) Repair in Giant Interphase and MS Cells is Associated with Promyelocytic Leukemia (PML) Bodies and is Featured by Alternative Lengthening of Telomeres (ALT)
2.7. Telomere Ends are Released into the Cytoplasm by MS
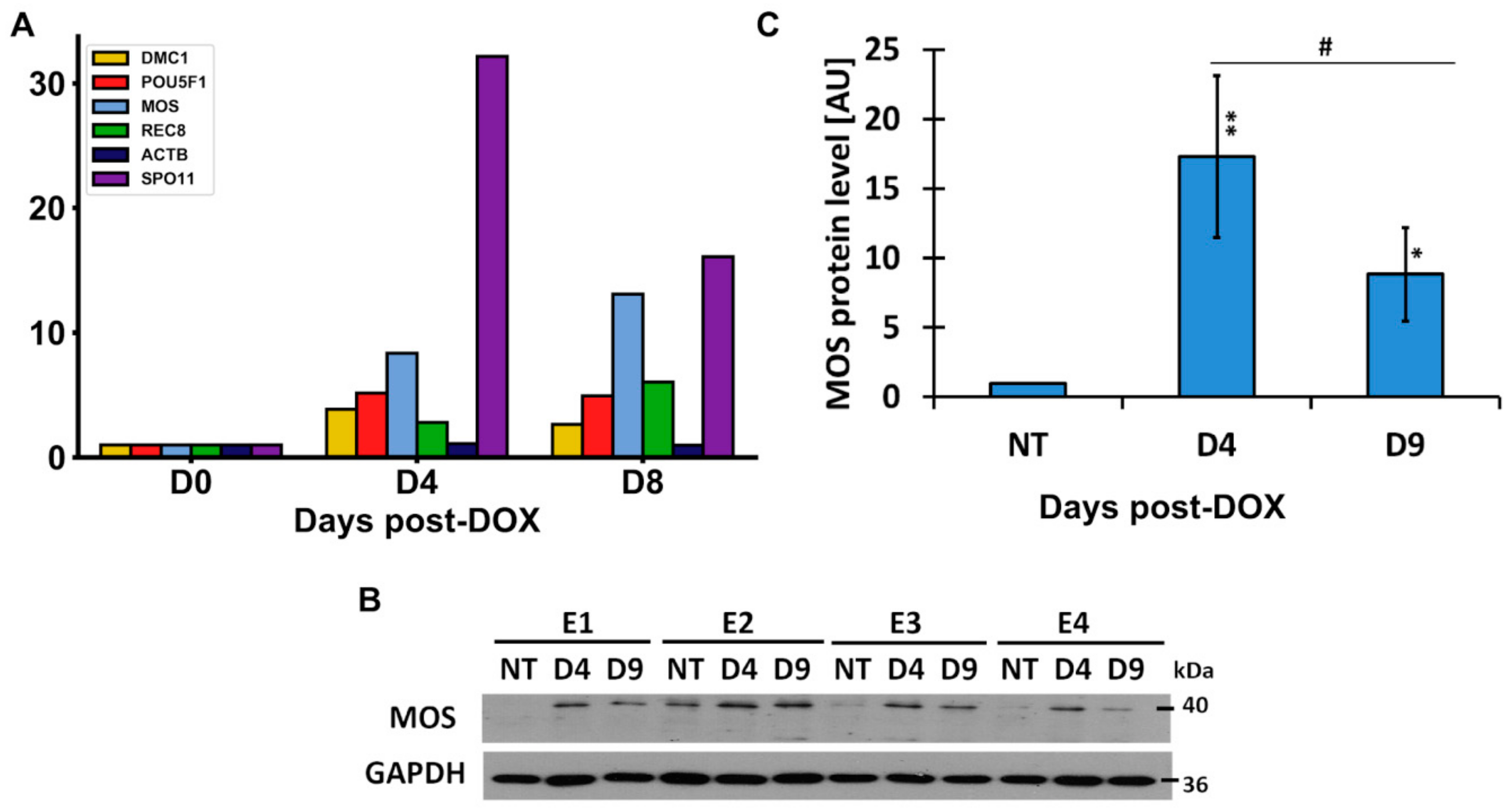
2.8. The Meiotic Features of “Mitotic Slippage”
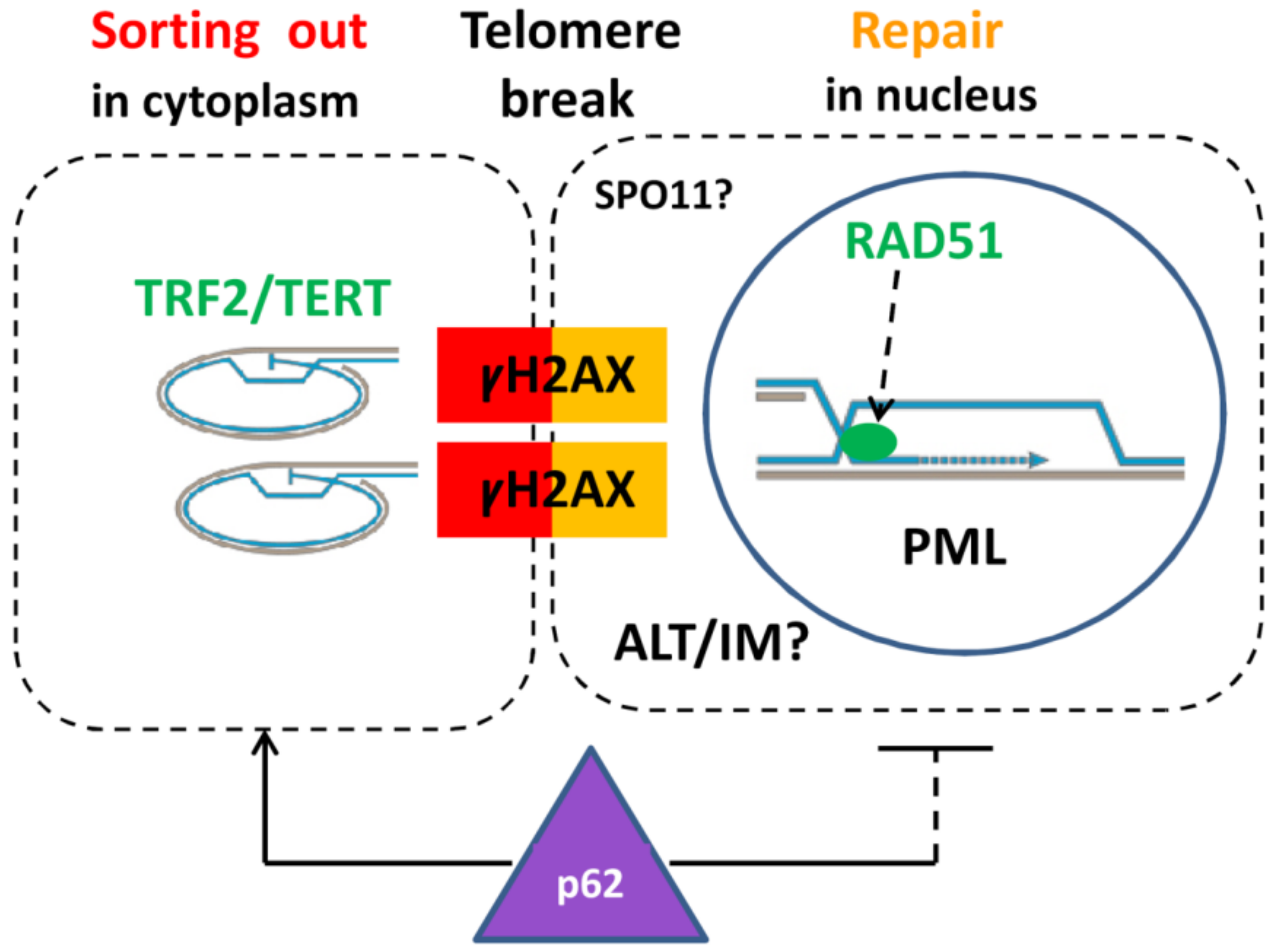
2.9. Extranuclear Sorting of the Cut-Off Telomere Ends in MS Being Regulated with the Participation of the Polyubiquitination Adaptor Protein SQSTM1/p62
3. Discussion
4. Materials and Methods
4.1. Cell Line and Treatment
4.2. Cell Growth, Viability, and Colony Formation
4.3. Immunofluorescence
4.4. Toluidine Blue DNA Staining and Image Cytometry
4.5. Fluorescence in Situ Hybridization (FISH)
4.6. Detection of Senescence-Associated-β-Galactosidase
4.7. RT-PCR
4.8. Selfie-Digital RT-PCR
4.9. Western Blot Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DOX | Doxorubicin |
| MS | Mitotic slippage |
| IM | Inverted meiosis |
| HR | Homologous recombination |
| PML | Promyelocytic leukemia (protein) |
| DSB | Double-strand break |
| ALT | Alternative telomere lengthening |
| TMM | Telomere maintenance mechanism |
| EMT | Epithelial–mesenchymal transition |
| NT | Non-treated |
| FBS | Fetal bovine serum |
References
- Riffell, J.L.; Zimmerman, C.; Khong, A.; McHardy, L.M.; Roberge, M. Effects of chemical manipulation of mitotic arrest and slippage on cancer cell survival and proliferation. Cell Cycle 2009, 8, 3025–3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Illidge, T.M.; Cragg, M.S.; Fringes, B.; Olive, P.; Erenpreisa, J.A. Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol. Int. 2000, 24, 621–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaram, M.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, R. Neosis: A novel type of cell division in cancer. Cancer Biol. Ther. 2004, 3, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Cragg, M.S. Cancer: A matter of life cycle? Cell Biol. Int. 2007, 31, 1507–1510. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Mercado-Uribe, I.; Xing, Z.; Sun, B.; Kuang, J.; Liu, J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene 2014, 33, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Carballo, D.; Gustmann, S.; Jastrow, H.; Acikelli, A.H.; Dammann, P.; Klein, J.; Dembinski, U.; Bardenheuer, W.; Malak, S.; Araúzo-Bravo, M.J.; et al. Atypical cell populations associated with acquired resistance to cytostatics and cancer stem cell features: The role of mitochondria in nuclear encapsulation. DNA Cell Biol. 2014, 33, 749–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Carballo, D.; Saka, S.; Klein, J.; Rennkamp, T.; Acikelli, A.H.; Malak, S.; Jastrow, H.; Wennemuth, G.; Tempfer, C.; Schmitz, I.; et al. A Distinct Oncogenerative Multinucleated Cancer Cell Serves as a Source of Stemness and Tumor Heterogeneity. Cancer Res. 2018, 78, 2318–2331. [Google Scholar] [CrossRef] [Green Version]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [Green Version]
- Salmina, K.; Jankevics, E.; Huna, A.; Perminov, D.; Radovica, I.; Klymenko, T.; Ivanov, A.; Jascenko, E.; Scherthan, H.; Cragg, M.; et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp. Cell Res. 2010, 316, 2099–2112. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmiņa, K.; Belyayev, A.; Inashkina, I.; Cragg, M.S. Survival at the Brink. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2017; pp. 275–294. ISBN 9780128121467. [Google Scholar]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Kumar, P.; Murray, D. Multinucleated Giant Cancer Cells Produced in Response to Ionizing Radiation Retain Viability and Replicate Their Genome. Int. J. Mol. Sci. 2017, 18, 360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid Giant Cancer Cells (PGCCs): The Evil Roots of Cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Krämer, A.; Hehlgans, S.; Sanhaji, M.; Kurunci-Csacsko, E.; Dötsch, C.; Bug, G.; Ottmann, O.; Becker, S.; Pachl, F.; et al. Mitotic arrest and slippage induced by pharmacological inhibition of Polo-like kinase 1. Mol. Oncol. 2015, 9, 140–154. [Google Scholar] [CrossRef]
- Rajaraman, R.; Guernsey, D.L.; Rajaraman, M.M.; Rajaraman, S.R. Stem cells, senescence, neosis and self-renewal in cancer. Cancer Cell Int. 2006, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Puig, P.-E.; Guilly, M.-N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Sliwinska, M.A.; Mosieniak, G.; Wolanin, K.; Babik, A.; Piwocka, K.; Magalska, A.; Szczepanowska, J.; Fronk, J.; Sikora, E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech. Ageing Dev. 2009, 130, 24–32. [Google Scholar] [CrossRef]
- Huna, A.; Salmina, K.; Erenpreisa, J.; Vazquez-Martin, A.; Krigerts, J.; Inashkina, I.; Gerashchenko, B.I.; Townsend, P.A.; Cragg, M.S.; Jackson, T.R. Role of stress-activated OCT4A in the cell fate decisions of embryonal carcinoma cells treated with etoposide. Cell Cycle 2015, 14, 2969–2984. [Google Scholar] [CrossRef]
- Sikora, E.; Mosieniak, G.; Sliwinska, M.A. Morphological and Functional Characteristic of Senescent Cancer Cells. Curr. Drug Targets 2016, 17, 377–387. [Google Scholar] [CrossRef]
- Leikam, C.; Hufnagel, A.; Walz, S.; Kneitz, S.; Fekete, A.; Müller, M.J.; Eilers, M.; Schartl, M.; Meierjohann, S. Cystathionase mediates senescence evasion in melanocytes and melanoma cells. Oncogene 2014, 33, 771–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Was, H.; Barszcz, K.; Czarnecka, J.; Kowalczyk, A.; Bernas, T.; Uzarowska, E.; Koza, P.; Klejman, A.; Piwocka, K.; Kaminska, B.; et al. Bafilomycin A1 triggers proliferative potential of senescent cancer cells in vitro and in NOD/SCID mice. Oncotarget 2017, 8, 9303–9322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosteiro, L.; Pantoja, C.; de Martino, A.; Serrano, M. Senescence promotes in vivo reprogramming through p16 and IL-6. Aging Cell 2018, 17, e12711. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerashchenko, B.I.; Salmina, K.; Eglitis, J.; Huna, A.; Grjunberga, V.; Erenpreisa, J. Disentangling the aneuploidy and senescence paradoxes: A study of triploid breast cancers non-responsive to neoadjuvant therapy. Histochem. Cell Biol. 2016, 145, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Gerashchenko, B.I.; Salmina, K.; Krigerts, J.; Erenpreisa, J.; Babsky, A.M. Induced polyploidy and sorting of damaged DNA by micronucleation in radioresistant rat liver epithelial stem-like cells exposed to X-rays. Probl. Radiac. Med. Radiobiol. 2019, 24, 220–234. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [Google Scholar] [CrossRef] [Green Version]
- Pluquet, O.; Abbadie, C.; Coqueret, O. Connecting cancer relapse with senescence. Cancer Lett. 2019, 463, 50–58. [Google Scholar] [CrossRef]
- Krtolica, A.; Campisi, J. Cancer and aging: A model for the cancer promoting effects of the aging stroma. Int. J. Biochem. Cell Biol. 2002, 34, 1401–1414. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Salmina, K.; Cragg, M.S. Accelerated Senescence of Cancer Stem Cells: A Failure to Thrive or a Route to Survival? In Senescence Physiology or Pathology; Dorszewska, J., Kozubski, W., Eds.; InTechOpen: London, UK, 2017; ISBN 9789535134619. [Google Scholar]
- Bharadwaj, D.; Mandal, M. Senescence in polyploid giant cancer cells: A road that leads to chemoresistance. Cytokine Growth Factor Rev. 2020, 52, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Amend, S.R.; Torga, G.; Lin, K.-C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.-A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Y.Y.; Londoño, D.; Bouley, R.; Rooney, M.S.; Hacohen, N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 2014, 9, 180–192. [Google Scholar] [CrossRef] [Green Version]
- Lan, Y.Y.; Heather, J.M.; Eisenhaure, T.; Garris, C.S.; Lieb, D.; Raychowdhury, R.; Hacohen, N. Extranuclear DNA accumulates in aged cells and contributes to senescence and inflammation. Aging Cell 2019, 18, e12901. [Google Scholar] [CrossRef] [Green Version]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Jakhar, R.; Luijten, M.N.H.; Wong, A.X.F.; Cheng, B.; Guo, K.; Neo, S.P.; Au, B.; Kulkarni, M.; Lim, K.J.; Maimaiti, J.; et al. Autophagy Governs Protumorigenic Effects of Mitotic Slippage-induced Senescence. Mol. Cancer Res. 2018, 16, 1625–1640. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Erenpreisa, J.; Cragg, M.S.; Salmina, K.; Hausmann, M.; Scherthan, H. The role of meiotic cohesin REC8 in chromosome segregation in gamma irradiation-induced endopolyploid tumour cells. Exp. Cell Res. 2009, 315, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Ianzini, F.; Kosmacek, E.A.; Nelson, E.S.; Napoli, E.; Erenpreisa, J.; Kalejs, M.; Mackey, M.A. Activation of meiosis-specific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Res. 2009, 69, 2296–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Noh, K.H.; Chung, J.-Y.; Takikita, M.; Chung, E.J.; Kim, B.W.; Hewitt, S.M.; Kim, T.W.; Kim, J.-H. Synaptonemal complex protein 3 is a prognostic marker in cervical cancer. PLoS ONE 2014, 9, e98712. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.F.; Byrnes, D.M.; Eller, M.S.; Rosa, A.M.; Dabas, N.; Escandon, J.; Grichnik, J.M. Potential role of meiosis proteins in melanoma chromosomal instability. J. Skin Cancer 2013, 2013, 190109. [Google Scholar] [CrossRef] [Green Version]
- Yant, L.; Bomblies, K. Genome management and mismanagement--cell-level opportunities and challenges of whole-genome duplication. Genes Dev. 2015, 29, 2405–2419. [Google Scholar] [CrossRef] [Green Version]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef] [Green Version]
- Walen, K.H. Spontaneous cell transformation: Karyoplasts derived from multinucleated cells produce new cell growth in senescent human epithelial cell cultures. In Vitro Cell. Dev. Biol. Anim. 2004, 40, 150–158. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Life-Cycle Features of Tumour Cells. In Evolutionary Biology from Concept to Application; Pontarotti, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 75, pp. 61–71. ISBN 9783540789925. [Google Scholar]
- Fais, S.; Fauvarque, M.-O. TM9 and cannibalism: How to learn more about cancer by studying amoebae and invertebrates. Trends Mol. Med. 2012, 18, 4–5. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Giuliani, A.; Vinogradov, A.E.; Anatskaya, O.V. Stress-induced polyploidy shifts somatic cells towards a pro-tumourogenic unicellular gene transcription network. Cancer Hypotheses 2018, 1, 1–20, CH-ErenpreisaFinal2-DWFE.pdf. [Google Scholar]
- Hecht, I.; Bar-El, Y.; Balmer, F.; Natan, S.; Tsarfaty, I.; Schweitzer, F.; Ben-Jacob, E. Tumor invasion optimization by mesenchymal-amoeboid heterogeneity. Sci. Rep. 2015, 5, 10622. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, J.; Zhang, X.; McKee, D.; Wang, Z.-B.; Li, R.; Ma, C.; Sun, Q.-Y.; Liu, X.J. The small GTPase Cdc42 promotes membrane protrusion during polar body emission via ARP2-nucleated actin polymerization. Mol. Hum. Reprod. 2011, 17, 305–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamao, M.; Naoki, H.; Kunida, K.; Aoki, K.; Matsuda, M.; Ishii, S. Distinct predictive performance of Rac1 and Cdc42 in cell migration. Sci. Rep. 2015, 5, 17527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- Davoli, T.; Denchi, E.L.; de Lange, T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell 2010, 141, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Davoli, T.; de Lange, T. Telomere-driven tetraploidization occurs in human cells undergoing crisis and promotes transformation of mouse cells. Cancer Cell 2012, 21, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Chung, I.; Osterwald, S.; Deeg, K.I.; Rippe, K. PML body meets telomere: The beginning of an ALTernate ending? Nucleus 2012, 3, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Andreyeva, E.N.; Kolesnikova, T.D.; Belyaeva, E.S.; Glaser, R.L.; Zhimulev, I.F. Local DNA underreplication correlates with accumulation of phosphorylated H2Av in the Drosophila melanogaster polytene chromosomes. Chromosome Res. 2008, 16, 851–862. [Google Scholar] [CrossRef]
- Glover, L.; Alsford, S.; Horn, D. DNA break site at fragile subtelomeres determines probability and mechanism of antigenic variation in African trypanosomes. PLoS Pathog. 2013, 9, e1003260. [Google Scholar] [CrossRef]
- Arnoult, N.; Karlseder, J. ALT telomeres borrow from meiosis to get moving. Cell 2014, 159, 11–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hewitt, G.; Carroll, B.; Sarallah, R.; Correia-Melo, C.; Ogrodnik, M.; Nelson, G.; Otten, E.G.; Manni, D.; Antrobus, R.; Morgan, B.A.; et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016, 12, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. The reproductive life cycle of cancer: Hypotheses of cell of origin, TP53 drivers and stem cell conversions in the light of the atavistic cancer cell theory. Med. Hypotheses 2019, 123, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, V.F. Carcinogenesis: Recent Insights in Protist Stem Cell Biology Lead To a Better Understanding of Atavistic Mechanisms Implied in Cancer Development. MOJ Tumor Res. 2018, 1, 00004. [Google Scholar] [CrossRef]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Shi, G.; Zhang, L.; Li, F.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, 32280. [Google Scholar] [CrossRef]
- Dilley, R.L.; Greenberg, R.A. ALTernative Telomere Maintenance and Cancer. Trends Cancer Res. 2015, 1, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Li, L.; Zhang, D.; Wu, K.; Chen, Y.; Zeng, J.; Wang, X.; He, D. Twisted epithelial-to-mesenchymal transition promotes progression of surviving bladder cancer T24 cells with hTERT-dysfunction. PLoS ONE 2011, 6, e27748. [Google Scholar] [CrossRef] [Green Version]
- Yong, J.W.Y.; Yeo, X.; Khan, M.M.; Lee, M.B.; Hande, M.P. Stable expression of promyelocytic leukaemia (PML) protein in telomerase positive MCF7 cells results in alternative lengthening of telomeres phenotype. Genome Integr. 2012, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Pickett, H.A.; Cesare, A.J.; Johnston, R.L.; Neumann, A.A.; Reddel, R.R. Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 2009, 28, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Kalejs, M.; Erenpreisa, J. Cancer/testis antigens and gametogenesis: A review and “brain-storming” session. Cancer Cell Int. 2005, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalejs, M.; Ivanov, A.; Plakhins, G.; Cragg, M.S.; Emzinsh, D.; Illidge, T.M.; Erenpreisa, J. Upregulation of meiosis-specific genes in lymphoma cell lines following genotoxic insult and induction of mitotic catastrophe. BMC Cancer 2006, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Cragg, M.S. MOS, aneuploidy and the ploidy cycle of cancer cells. Oncogene 2010, 29, 5447–5451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “virgin birth”, polyploidy, and the origin of cancer. Oncoscience 2015, 2, 3–14. [Google Scholar] [CrossRef]
- Niu, N.; Mercado-Uribe, I.; Liu, J. Dedifferentiation into blastomere-like cancer stem cells via formation of polyploid giant cancer cells. Oncogene 2017, 36, 4887–4900. [Google Scholar] [CrossRef] [Green Version]
- Liu, J. The dualistic origin of human tumors. Semin. Cancer Biol. 2018, 53, 1–16. [Google Scholar] [CrossRef]
- Old, L.J. Cancer/testis (CT) antigens—A new link between gametogenesis and cancer. Cancer Immun. 2001, 1, 1. [Google Scholar]
- Erenpreisa, J.; Kalejs, M.; Cragg, M.S. Mitotic catastrophe and endomitosis in tumour cells: An evolutionary key to a molecular solution. Cell Biol. Int. 2005, 29, 1012–1018. [Google Scholar] [CrossRef]
- McFarlane, R.J.; Wakeman, J.A. Meiosis-like Functions in Oncogenesis: A New View of Cancer. Cancer Res. 2017, 77, 5712–5716. [Google Scholar] [CrossRef] [Green Version]
- Hofstatter, P.G.; Lahr, D.J.G. All Eukaryotes Are Sexual, unless Proven Otherwise: Many So-Called Asexuals Present Meiotic Machinery and Might Be Able to Have Sex. Bioessays 2019, 41, 1800246. [Google Scholar] [CrossRef]
- Forche, A.; Alby, K.; Schaefer, D.; Johnson, A.D.; Berman, J.; Bennett, R.J. The parasexual cycle in Candida albicans provides an alternative pathway to meiosis for the formation of recombinant strains. PLoS Biol. 2008, 6, e110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viera, A.; Page, J.; Rufas, J.S. Inverted meiosis: The true bugs as a model to study. Genome Dyn. 2009, 5, 137–156. [Google Scholar] [PubMed]
- Kuznetsova, V.G. The Chromosomes of the Holokinetic Type and Their Distribution among Insects and Other Invertebrate Animals. In Kariosistematika Bespozvonochnykh Zhivotnykh (Karyosystematics of Invertebrates); Nauka: Leningrad, Russia, 1979; pp. 5–19. [Google Scholar]
- Solari, A.J. Primitive forms of meiosis: The possible evolution of meiosis. Biocell 2002, 26, 1–13. [Google Scholar]
- Heckmann, S.; Jankowska, M.; Schubert, V.; Kumke, K.; Ma, W.; Houben, A. Alternative meiotic chromatid segregation in the holocentric plant Luzula elegans. Nat. Commun. 2014, 5, 4979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanov, Y.F. Inverted meiosis and its place in the evolution of sexual reproduction pathways. Russ. J. Genet. 2016, 52, 473–490. [Google Scholar] [CrossRef]
- Loidl, J. Conservation and Variability of Meiosis Across the Eukaryotes. Annu. Rev. Genet. 2016, 50, 293–316. [Google Scholar] [CrossRef]
- Ottolini, C.S.; Newnham, L.; Capalbo, A.; Natesan, S.A.; Joshi, H.A.; Cimadomo, D.; Griffin, D.K.; Sage, K.; Summers, M.C.; Thornhill, A.R.; et al. Genome-wide maps of recombination and chromosome segregation in human oocytes and embryos show selection for maternal recombination rates. Nat. Genet. 2015, 47, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Ottolini, C.S. Chromosome Segregation and Recombination in Human Meiosis: Clinical Applications and Insight Into Disjunction Errors. Ph.D. Thesis, University of Kent, Canterbury, UK, 2015. Doctor of Philosophy. [Google Scholar]
- Perrem, K.; Colgin, L.M.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Coexistence of alternative lengthening of telomeres and telomerase in hTERT-transfected GM847 cells. Mol. Cell. Biol. 2001, 21, 3862–3875. [Google Scholar] [CrossRef] [Green Version]
- Chiodi, I.; Mondello, C. Telomere-independent functions of telomerase in nuclei, cytoplasm, and mitochondria. Front. Oncol. 2012, 2, 133. [Google Scholar] [CrossRef] [Green Version]
- Maciver, S.K. Asexual Amoebae Escape Muller’s Ratchet through Polyploidy. Trends Parasitol. 2016, 32, 855–862. [Google Scholar] [CrossRef]
- Khan, N.A.; Siddiqui, R. Is there evidence of sexual reproduction (meiosis) in Acanthamoeba? Pathog. Glob. Health 2015, 109, 193–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demin, S.Y.; Berdieva, M.A.; Goodkov, A.V. Cyclic Polyploidy in Obligate Agamic Amoebae. Cell Tissue Biol. 2019, 13, 242–246. [Google Scholar] [CrossRef]
- Goodkov, A.V.; Berdieva, M.A.; Podlipaeva, Y.I.; Demin, S.Y. The Chromatin Extrusion Phenomenon in Amoeba proteus Cell Cycle. J. Eukaryot. Microbiol. 2019, 67, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Berdieva, M.; Demin, S.; Goodkov, A. Amoeba proteus and ploidy cycles: From simple model to complex issues. Protistology 2019, 13, 166–173. [Google Scholar] [CrossRef]
- Archetti, M. Inverted meiosis and the evolution of sex by loss of complementation. J. Evol. Biol. 2019. [Google Scholar] [CrossRef]
- Zybina, T.G. Genome variation in the trophoblast cell lifespan: Diploidy, polyteny, depolytenization, genome segregation. WJMG 2014, 4, 77. [Google Scholar] [CrossRef]
- Zybina, T.G.; Stein, G.I.; Pozharisski, K.M.; Zybina, E.V. Invasion and genome reproduction of the trophoblast cells of placenta junctional zone in the field vole, Microtus rossiaemeridionalis. Cell Biol. Int. 2014, 38, 136–143. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Jackson, T.R.; Salmina, K.; Huna, A.; Inashkina, I.; Jankevics, E.; Riekstina, U.; Kalnina, Z.; Ivanov, A.; Townsend, P.A.; Cragg, M.S.; et al. DNA damage causes TP53-dependent coupling of self-renewal and senescence pathways in embryonal carcinoma cells. Cell Cycle 2013, 12, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Narath, R.; Lörch, T.; Greulich-Bode, K.M.; Boukamp, P.; Ambros, P.F. Automatic telomere length measurements in interphase nuclei by IQ-FISH. Cytometry A 2005, 68, 113–120. [Google Scholar] [CrossRef]
- Scherthan, H.; Sotnik, N.; Peper, M.; Schrock, G.; Azizova, T.; Abend, M. Telomere Length in Aged Mayak PA Nuclear Workers Chronically Exposed to Internal Alpha and External Gamma Radiation. Radiat. Res. 2016, 185, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Asadi, M.H.; Khalifeh, K.; Mowla, S.J. OCT4 spliced variants are highly expressed in brain cancer tissues and inhibition of OCT4B1 causes G2/M arrest in brain cancer cells. J. Neurooncol. 2016, 130, 455–463. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [Green Version]
- Podlesniy, P.; Trullas, R. Absolute measurement of gene transcripts with Selfie-digital PCR. Sci. Rep. 2017, 7, 8328. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Average Ploidy * | Normal Cycle | Aberrant Metaphase | Mitotic Slippage | Polyploidy Cycles | Hyperploidy > 20C | Normal Mitosis |
|---|---|---|---|---|---|---|---|
| Day 0 | 2.68C | + | − | − | − | − | + |
| Day 4 | 3.65C | − 1 | ++ | + | + | − | − |
| Day 8 | 7.47C | − | + | ++ | +++ | + | − |
| Day 16 | 12.15C | +/− | +/− | +++ | ++ | +++ | - |
| Day 18 | 8.87C | + | − | + | + | ++ | + |
| Day 25 | 3.26C | + | − | − | − | −/+ | ++ |
| Antibody Against | Description | Specificity/Immunogen | Used Concentration | Product No. and Manufacturer |
|---|---|---|---|---|
| AURORA B | Rabbit polyclonal | A peptide derived from within residues 1–100 of human Aurora B | 1:300 | ab2254, Abcam, Cambrige, UK |
| BrdU | Mouse monoclonal | Recognizes the thymidine analog 5-bromo-2′-deoxyuridine (BrdU) | 1:100 | A21300, Invitrogen, Carlsbad, CA, USA |
| α-Tubulin | Mouse monoclonal | Recognizes an epitope located at the C-terminal end of the α-tubulin isoform in a variety of organisms | 1:1000 | T5168, Sigma-Aldrich, St. Louis, MO, USA |
| Centromere protein | Human | Derived from human CREST patient serum | 1:50 | 15–234, Antibodies Inc., Davis, CA, USA |
| CYCLIN B1 | Mouse monoclonal | Raised against a recombinant protein corresponding to human cyclin B1 | 1:100 | sc-245, Santa Cruz, Dallas, TX, USA |
| DMC1 | Mouse monoclonal | Specific for DMC1, does not cross-react with the related protein Rad51 | 1:100 | ab11054, Abcam, Cambridge, UK |
| F-ACTIN | Phalloidin-iFlour 594 Conjugate | 1:500 | ab176757, Abcam, Cambridge, UK | |
| FRAGILIS | Rabbit polyclonal | The details of the immunogen for this antibody are not available | 1:50 | ab65183-100, Abcam, Cambridge, UK |
| GAPDH | Mouse monoclonal | Raised against recombinant GAPDH of human origin | 1:50000 | sc-47724, Santa Cruz, Dallas, TX, USA |
| IL6 | Rabbit polyclonal | Synthetic peptide | 1:50 | orb87798, Biorbyt, Cambridge, UK |
| γ-H2AX | Rabbit polyclonal | Recognizes mammalian, yeast Drosophila melanogaster and Xenopus laevis γ-H2AX | 1:200 | 4411-PC-100, Trevigen, Gaithersburg, MD, USA |
| γ-H2AX | Mouse monoclonal | Synthetic peptide sequence surrounding phosphorylated Ser140 | 1:200 | Ma1-2022, Pierce, Waltham, MA, USA |
| H3K27me3 | Rabbit polyclonal | Synthetic peptide within human Histone H3 aa 1–100 | 1:200 | ab6147, Abcam, Cambridge, UK |
| Ki67 | Rabbit polyclonal | Synthetic peptide from C-terminus of human Ki-67 | 1:50 | PA5-16785, Pierce, Waltham, MA, USA |
| LAMIN B1 | Rabbit polyclonal | Peptide mapping at the C-terminus of Lamin B1 of human origin | 1:200 | ab1604, Abcam, Cambridge, UK |
| LAMP2 | Mouse monoclonal | The details of the immunogen for this antibody are not available | 1:500 | 555803, BD Pharmingen™, Franklin Lakes, NJ, USA |
| MOS (C237) | Rabbit polyclonal | Epitope mapping at the C-terminus | 1:50 | sc-86, Santa Cruz, Dallas, TX, USA |
| MOS | Rabbit polyclonal | Synthetic peptide corresponding to a region within internal sequence amino acids 107–156 | 1:500 | Ab99017, Abcam, Cambridge, UK |
| OCT 3/4 | Mouse monoclonal | Peptide raised against amino acids 1–134 of Oct-3/4 of human origin non-cross-reactive with Oct-3/4 isoforms B and B1 | 1:50 | sc-5279, Santa Cruz, Dallas, TX, USA |
| OCT4 | Rabbit polyclonal | A peptide derived from within residues 300 to the C-terminus of human Oct4 | 1:200 | ab19857, Abcam, Cambridge, UK |
| p-AMPKα1/2 (Thr183/172) | Rabbit polyclonal | Epitope corresponding to phosphorylated Thr172 of AMPKα1 of human origin | 1:50 | sc-101630, Santa Cruz, Dallas, TX, USA |
| pH3Ser10 | Mouse monoclonal | Recognizes Phospho- S10 on Histone H3 | 1:200 | ab14955, Abcam, Cambridge, UK |
| PML | Mouse monoclonal | Epitope corresponding to amino acids 37–51 mapping near the N-terminal of PML of human origin | 1:200 | sc-966, Santa Cruz, Dallas, TX, USA |
| P62/SQSTM1 | Rabbit polyclonal | A synthetic peptide corresponding to Human SQSTM1/ p62 (C-terminal) | 1:200 | ab91526, Abcam, Cambridge, UK |
| RAD51 | Mouse monoclonal | Recombinant full-length protein corresponding to human Rad51 aa 1–338 | 1:50 | ab213, Abcam, Cambridge, UK |
| REC8 | Rabbit polyclonal | Peptide mapping near the N-terminus of Rec8 of human origin | 1:50 | 10793-1-AP, Proteintech Group, Manchester, UK |
| TERT | Mouse monoclonal | Recombinant full-length protein (human) from insect cells | 1:50 | ab5181, Abcam, Cambridge, UK |
| TRF2 | Mouse monoclonal | His-tagged, fusion protein, corresponding to full-length TRF2 (Telomeric Repeat binding Factor 2) | 1:100 | 05-521, Millipore, Temecula, CA, USA |
| VASA/DDX4 | Mouse monoclonal | A synthetic peptide corresponding to residues near the N-terminus of human DDX4 | 1:50 | MA5-15565, Pierce, Waltham, MA, USA |
| 5-Methylcytosine | Mouse monoclonal | Detects methylated DNA or RNA | 1:200 | NA81, Calbiochem, Merck, Burlington, MA, USA |
| Gene | Forward Primer Sequence | Reverse Primer Sequence | Amplicon Length | Tann (°C) | Reference | Sequence ID |
|---|---|---|---|---|---|---|
| POU5F1-A (Oct4A) | TCGCAAGCCCTCATTTCACC | GCCAGGTCCGAGGATCAAC | 157 | 56 | [109] | NM_002701.5 |
| POU5F1-B (Oct4B) | AGACTATTCCTTGGGGCCACAC | GGCTGAATACCTTCCCAAATAGA | 244 | 58 | [109] | NM_203289.5 |
| POU5F1-B1(Oct4B1) | TGACCGCATCTCCCCTCTAA | AGCTTACCACCTCTTCCCAG | 134 | 58 | [109] | NM_001285986.1 |
| MOS | CGGTGTTCCTGTGGCCATAA | GCAGGCCGTTCACAACATC | 250 | 58 | [75] | NM_005372.1 |
| REC8 | TGAGGGTGAATGTGGTGAAA | CTGGGATTGCAGCCTCTAAG | 400 | 56 | [75] | NM_005132.2 |
| DMC1 | AGCAGCAAAGTTCCATGAAG | TGAGCTCTCCTCTTCCCTTT | 300 | 54 | [75] | NM_007068.3 |
| SPO11 | TGAGGTTCTTGCATCTATAGAAA | AAATTTTTGAGCTGATTTTGGTG | 240 | 58 | in house | NM_012444.2 |
| ACTB | AGTGTGACGTGGACATCCG | AATCTCATCTTGTTTTCTGCGC | 349 | 56 | [110] | NA |
| Gene Symbol | Amplicon Size | Forward Primer Sequence 5′-3′ | Reverse Primer Sequence 5′-3′ |
|---|---|---|---|
| RAC-1 | 75 bp. | AAACCGGTGAATCTGGGCTT | CGGATAGGATAGGGGGCGTA |
| POU5F1 | 97 bp. | GAGTAGTCCCTTCGCAAGCC | GAGAAGGCGAAATCCGAAGC |
| CDC-42 | 95 bp. | TGTTGAACCAATGCTTTCTCATGT | CTCAGGCTGGCTTGTGAAGG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmina, K.; Bojko, A.; Inashkina, I.; Staniak, K.; Dudkowska, M.; Podlesniy, P.; Rumnieks, F.; Vainshelbaum, N.M.; Pjanova, D.; Sikora, E.; et al. “Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres. Int. J. Mol. Sci. 2020, 21, 2779. https://doi.org/10.3390/ijms21082779
Salmina K, Bojko A, Inashkina I, Staniak K, Dudkowska M, Podlesniy P, Rumnieks F, Vainshelbaum NM, Pjanova D, Sikora E, et al. “Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres. International Journal of Molecular Sciences. 2020; 21(8):2779. https://doi.org/10.3390/ijms21082779
Chicago/Turabian StyleSalmina, Kristine, Agnieszka Bojko, Inna Inashkina, Karolina Staniak, Magdalena Dudkowska, Petar Podlesniy, Felikss Rumnieks, Ninel M Vainshelbaum, Dace Pjanova, Ewa Sikora, and et al. 2020. "“Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres" International Journal of Molecular Sciences 21, no. 8: 2779. https://doi.org/10.3390/ijms21082779