Inflammatory and Infectious Processes Serve as Links between Atrial Fibrillation and Alzheimer’s Disease

Abstract
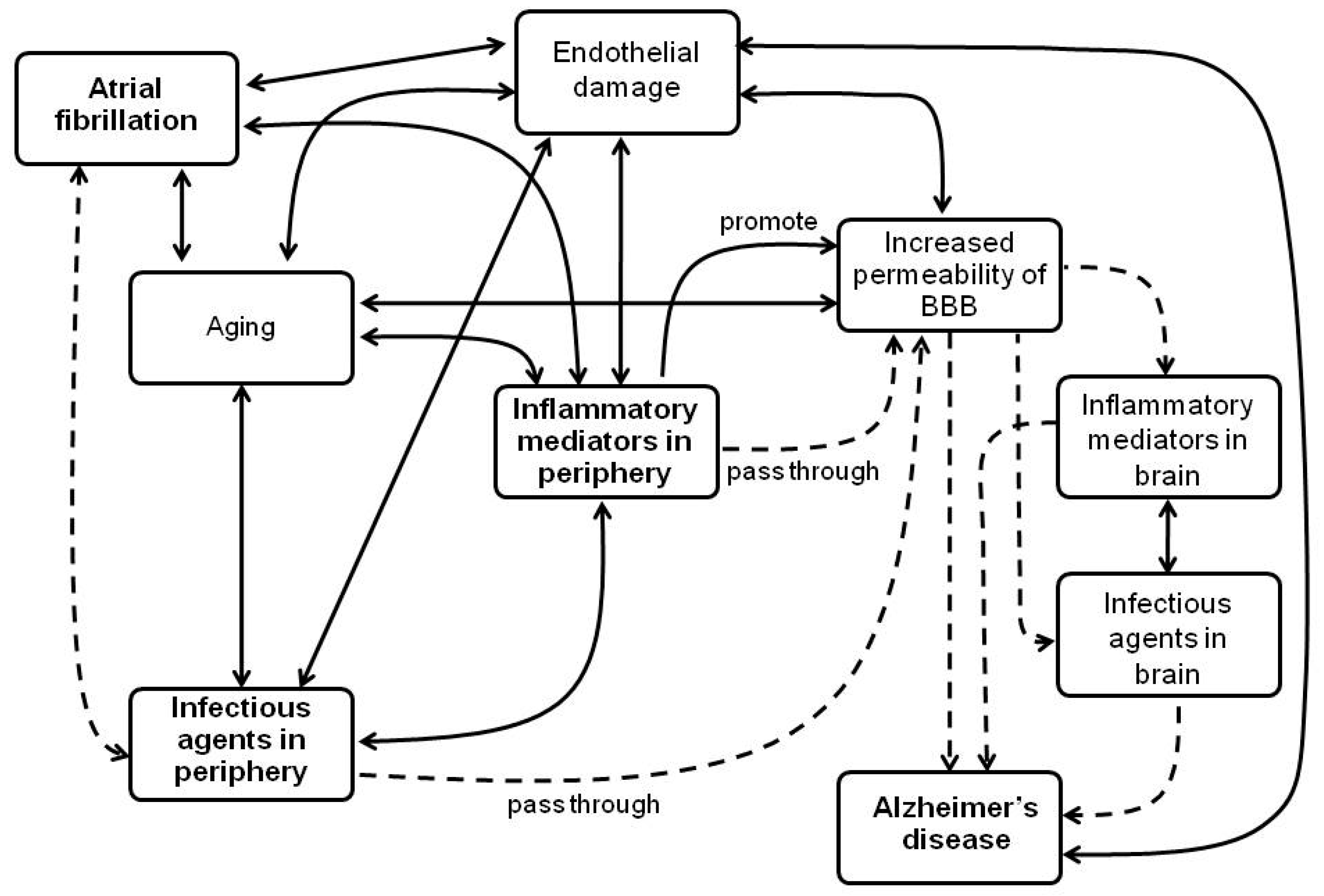
:1. Introduction
2. Inflammatory Mechanisms in AF and in AD, and Its Association between Both Diseases
2.1. Inflammatory Mediators Associated with Both AF and AD
2.2. Endothelial Damage Associated with Inflammation and Both AF and AD
3. Infectious Agents Related to AF and AD as a Possible Link between Both Diseases
4. Pharmacological Approaches
4.1. Anti-Inflammatory Therapies for AF and AD
4.1.1. Non-Steroidal Anti-Inflammatory Drugs and Corticosteroids
4.1.2. Agents That Modulate Cytokines Signaling
4.1.3. Other Drugs That Have Anti-Inflammatory Properties
4.2. Therapies for AF and Its Potential Role in Prevention of Dementia and AD
4.3. Paradigm in the Treatment of Patients With AD Who Have AF
5. Final Considerations
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACEIs | Angiotensin converting enzyme inhibitors |
| AD | Alzheimer’s disease |
| AEPU | 1-adamantan-1-yl-3-{5-[2-(2-ethoxy-ethoxy)-ethoxy]-pentyl}-urea |
| AF | Atrial fibrillation |
| ARBs | Angiotensin-1 receptor blockers |
| BBB | Blood brain barrier |
| BCSFB | Blood-cerebrospinal fluid barrier |
| CAIDE | Cardiovascular Risk Factors, Aging and Dementia |
| ChEI | Cholinesterase inhibitors |
| CI | Confidence interval |
| CNS | Central nervous system |
| COX | Cyclooxygenase |
| CRP | C reactive protein |
| CSF | Cerebrospinal fluid |
| EETs | Epoxy-eicosa-trienoic acids |
| EPHX2 | Epoxide hydrolase 2 |
| HF | Heart failure |
| HR | Hazard ratio |
| HSPs | Heat shock proteins |
| HSV | Herpes simplex virus |
| Ig | Immunoglobulin |
| IL | Interleukin |
| MCI | Mild cognition impairment |
| MCP | Monocyte chemo-attractant protein |
| MI | Myocardial infarction |
| mRNA | Messenger ribonucleic acid |
| NOACs | Novel oral anticoagulants |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| OACs | Oral anticoagulants |
| OR | Odds ratio |
| PUFAs | Poly-unsaturated fatty acids |
| RAAS | Renin-angiotensin-aldosterone system |
| RR | Relative risk |
| sEH | Soluble epoxide hydrolase |
| sEHI | Soluble epoxide hydrolase inhibitors |
| t-AUCB | trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid |
| TGF | Transforming growth factor |
| TNF | Tumor necrosis factor |
| TPPU | Trifluoro-methoxy-phenyl-3-(1-propionyl-piperidine-4-yl)-urea |
| βAP | β-amyloid peptide |
References
- Savelieva, I.; Kakouros, N.; Kourliouros, A.; Camm, A.J. Upstream therapies for management of atrial fibrillation: Review of clinical evidence and implications for European Society of Cardiology guidelines. Part I: Primary prevention. Europace 2011, 13, 308–328. [Google Scholar] [CrossRef]
- Vlachos, K.; Letsas, K.P.; Korantzopoulos, P.; Liu, T.; Georgopoulos, S.; Bakalakos, A.; Karamichalakis, N.; Xydonas, S.; Efremidis, M.; Sideris, A. Prediction of atrial fibrillation development and progression: Current perspectives. World J. Cardiol. 2016, 8, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Washida, K. Linking atrial fibrillation with alzheimer’s disease: Epidemiological, pathological, and mechanistic evidence. J. Alzheimers Dis. 2018, 62, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietzel, J.; Haeusler, K.G.; Endres, M. Does atrial fibrillation cause cognitive decline and dementia? EP Eur. 2017, 20, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Sposato, L.A.; Vargas, E.R.; Riccio, P.M.; Toledo, J.B.; Trojanowski, J.Q.; Kukull, W.A.; Cipriano, L.E.; Nucera, A.; Whitehead, S.N.; Hachinski, V. Milder Alzheimer’s disease pathology in heart failure and atrial fibrillation. Alzheimers Dement. 2017, 13, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Y.; Norby, F.L.; Gottesman, R.F.; Mosley, T.H.; Soliman, E.Z.; Agarwal, S.K.; Loehr, L.R.; Folsom, A.R.; Coresh, J.; Alonso, A. Association of Atrial Fibrillation with Cognitive Decline and Dementia Over 20 Years: The ARIC-NCS (Atherosclerosis Risk in Communities Neurocognitive Study). J. Am. Heart Assoc. 2018, 7, e007301. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Winblad, B.; Marengoni, A.; Klarin, I.; Fastbom, J.; Fratiglioni, L. Heart failure and risk of dementia and Alzheimer disease: A population-based cohort study. Arch. Intern. Med. 2006, 166, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Dublin, S.; Anderson, M.L.; Haneuse, S.J.; Heckbert, S.R.; Crane, P.K.; Breitner, J.C.; McCormick, W.; Bowen, J.D.; Teri, L.; McCurry, S.M. Atrial fibrillation and risk of dementia: A prospective cohort study. J. Am. Geriatr. Soc. 2011, 59, 1369–1375. [Google Scholar] [CrossRef] [Green Version]
- Rusanen, M.; Kivipelto, M.; Levälahti, E.; Laatikainen, T.; Tuomilehto, J.; Soininen, H.; Ngandu, T. Heart diseases and long-term risk of dementia and Alzheimer’s disease: A population-based CAIDE study. J. Alzheimers Dis. 2014, 42, 183–191. [Google Scholar] [CrossRef]
- Di Nisio, M.; Prisciandaro, M.; Rutjes, A.W.; Russi, I.; Maiorini, L.; Porreca, E. Dementia in patients with atrial fibrillation and the value of the H achinski ischemic score. Geriatr. Gerontol. Int. 2015, 15, 770–777. [Google Scholar] [CrossRef]
- Bunch, T.J.; Weiss, J.P.; Crandall, B.G.; May, H.T.; Bair, T.L.; Osborn, J.S.; Anderson, J.L.; Muhlestein, J.B.; Horne, B.D.; Lappe, D.L. Atrial fibrillation is independently associated with senile, vascular, and Alzheimer’s dementia. Heart Rhythm 2010, 7, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Knecht, S.; Oelschläger, C.; Duning, T.; Lohmann, H.; Albers, J.; Stehling, C.; Heindel, W.; Breithardt, G.; Berger, K.; Ringelstein, E.B. Atrial fibrillation in stroke-free patients is associated with memory impairment and hippocampal atrophy. Eur. Heart J. 2008, 29, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Dublin, S.; Anderson, M.L.; Heckbert, S.R.; Hubbard, R.A.; Sonnen, J.A.; Crane, P.K.; Montine, T.J.; Larson, E.B. Neuropathologic changes associated with atrial fibrillation in a population-based autopsy cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2013, 69, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSimone, C.V.; Graff-Radford, J.; El-Harasis, M.A.; Rabinstein, A.A.; Asirvatham, S.J.; Holmes, D.R. Cerebral amyloid angiopathy: Diagnosis, clinical implications, and management strategies in atrial fibrillation. J. Am. Coll. Cardiol. 2017, 70, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Qiu, C. Atrial Fibrillation, Cognitive Decline, and Dementia: An Epidemiologic Review. Curr. Epidemiol. Rep. 2018, 5, 252–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.D.; Merchant, F.M.; Delurgio, D.B. Atrial fibrillation and risk of dementia/cognitive decline. J. Atr. Fibrillation 2016, 8, 1353. [Google Scholar] [PubMed]
- Jefferson, A.L.; Beiser, A.S.; Himali, J.J.; Seshadri, S.; O’Donnell, C.J.; Manning, W.J.; Wolf, P.A.; Au, R.; Benjamin, E.J. Low cardiac index is associated with incident dementia and Alzheimer disease: The Framingham Heart Study. Circulation 2015, 131, 1333–1339. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- Troncone, L.; Luciani, M.; Coggins, M.; Wilker, E.H.; Ho, C.-Y.; Codispoti, K.E.; Frosch, M.P.; Kayed, R.; Del Monte, F. Aβ amyloid pathology affects the hearts of patients with Alzheimer’s disease: Mind the Heart. J. Am. Coll. Cardiol. 2016, 68, 2395–2407. [Google Scholar] [CrossRef]
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ. J. 2015, 79, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Korantzopoulos, P.; Letsas, K.P.; Tse, G.; Fragakis, N.; Goudis, C.A.; Liu, T. Inflammation and atrial fibrillation: A comprehensive review. J. Arrhythm. 2018, 34, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease—A brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 6, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358. [Google Scholar] [CrossRef] [PubMed]
- Andrew, P.; Montenero, A.S. Is there a link between atrial fibrillation and certain bacterial infections? J. Cardiovasc. Med. (Hagerstown) 2007, 8, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Zwolinska, K.; Leszek, J. The infectious etiology of Alzheimer’s disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [Green Version]
- Magalhães, L.P.; Figueiredo, M.J.O.; Cintra, F.D.; Saad, E.B.; Kuniyishi, R.R.; Teixeira, R.A. II Diretrizes Brasileiras de Fibrilação Atrial. Arq. Bras. Cardiol. 2016, 106, 1–22. [Google Scholar] [CrossRef]
- Hu, Y.F.; Chen, Y.J.; Lin, Y.J.; Chen, S.A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef]
- Watson, T.; Shantsila, E.; Lip, G.Y. Mechanisms of thrombogenesis in atrial fibrillation: Virchow’s triad revisited. Lancet 2009, 373, 155–166. [Google Scholar] [CrossRef]
- Ott, B.R.; Jones, R.; Daiello, L.A.; Monte, S.d.L.; Stopa, E.G.; Johanson, C.E.; Denby, C.; Grammas, P. Blood-Cerebrospinal Fluid Barrier Gradients in Mild Cognitive Impairment and Alzheimer’s Disease: Relationship to Inflammatory Cytokines and Chemokines. Front. Aging Neurosci. 2018, 10, 245. [Google Scholar] [CrossRef] [Green Version]
- Farrall, A.J.; Wardlaw, J.M. Blood–brain barrier: Ageing and microvascular disease–systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Johanson, C.; Stopa, E.; Daiello, L.; De la Monte, S.; Keane, M.; Ott, B. Disrupted blood-CSF barrier to urea and creatinine in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. Parkinsonism 2018, 8, 435. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.Y.; Kim, D.H.; Lee, E.K.; Chung, K.W.; Chung, S.; Lee, B.; Seo, A.Y.; Chung, J.H.; Jung, Y.S.; Im, E. Redefining Chronic Inflammation in Aging and Age-Related Diseases: Proposal of the Senoinflammation Concept. Aging Dis. 2019, 10, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elwood, E.; Lim, Z.; Naveed, H.; Galea, I. The effect of systemic inflammation on human brain barrier function. Brain. Behav. Immun. 2017, 62, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, L.Y.; Venneri, A.; Farkas, E.; Evans, P.C.; Marzo, A.; Frangi, A.F. Vascular dysfunction in the pathogenesis of Alzheimer’s disease—A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol. Dis. 2015, 82, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Sato, N.; Morishita, R. Systemic inflammation, blood-brain barrier vulnerability and cognitive/non-cognitive symptoms in Alzheimer disease: Relevance to pathogenesis and therapy. Front. Aging Neurosci. 2014, 6, 171. [Google Scholar] [CrossRef]
- Sun, Z.; Zhou, D.; Xie, X.; Wang, S.; Wang, Z.; Zhao, W.; Xu, H.; Zheng, L. Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res. Cardiol. 2016, 111, 63. [Google Scholar] [CrossRef] [Green Version]
- Kaireviciute, D.; Blann, A.D.; Balakrishnan, B.; Lane, D.A.; Patel, J.V.; Uzdavinys, G.; Norkunas, G.; Kalinauskas, G.; Sirvydis, V.; Aidietis, A.; et al. Characterisation and validity of inflammatory biomarkers in the prediction of post-operative atrial fibrillation in coronary artery disease patients. Thromb. Haemost. 2010, 104, 122–127. [Google Scholar] [CrossRef]
- Li, J.; Solus, J.; Chen, Q.; Rho, Y.H.; Milne, G.; Stein, C.M.; Darbar, D. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm 2010, 7, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Lee, K.J.; Kim, H. Serum tumour necrosis factor-α and interleukin-6 levels in Alzheimer’s disease and mild cognitive impairment. Psychogeriatrics 2017, 17, 224–230. [Google Scholar] [CrossRef]
- Paouri, E.; Tzara, O.; Zenelak, S.; Georgopoulos, S. Genetic Deletion of Tumor Necrosis Factor-α Attenuates Amyloid-β Production and Decreases Amyloid Plaque Formation and Glial Response in the 5XFAD Model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 165–181. [Google Scholar] [CrossRef] [PubMed]
- D’Anna, L.; Abu-Rumeileh, S.; Fabris, M.; Pistis, C.; Baldi, A.; Sanvilli, N.; Curcio, F.; Gigli, G.L.; D’Anna, S.; Valente, M. Serum interleukin-10 levels correlate with cerebrospinal fluid amyloid beta deposition in Alzheimer disease patients. Neurodegener Dis. 2017, 17, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Ke, D.; Xu, C.; Deng, Y.; Chen, L.; Zhang, J.; Lin, Y.; Hu, X. Collagen type I and Interleukin-1 beta gene expression in human atria during atrial fibrillation. Zhonghua Nei Ke Za Zhi 2006, 45, 807–810. [Google Scholar] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Forlenza, O.V.; Diniz, B.S.; Talib, L.L.; Mendonça, V.A.; Ojopi, E.B.; Gattaz, W.F.; Teixeira, A.L. Increased serum IL-1β level in Alzheimer’s disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2009, 28, 507–512. [Google Scholar] [CrossRef]
- Henningsen, K.M.; Therkelsen, S.K.; Bruunsgaard, H.; Krabbe, K.S.; Pedersen, B.K.; Svendsen, J.H. Prognostic impact of hs-CRP and IL-6 in patients with persistent atrial fibrillation treated with electrical cardioversion. Scand. J. Clin. Lab. Investig. 2009, 69, 425–432. [Google Scholar] [CrossRef]
- Hampel, H.; Haslinger, A.; Scheloske, M.; Padberg, F.; Fischer, P.; Unger, J.; Teipel, S.J.; Neumann, M.; Rosenberg, C.; Oshida, R. Pattern of interleukin-6 receptor complex immunoreactivity between cortical regions of rapid autopsy normal and Alzheimer’s disease brain. Eur. Arch. Psychiatry Clin. Neurosci. 2005, 255, 269–278. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Orellana, D.I.; González-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef]
- Pinto, A.; Tuttolomondo, A.; Casuccio, A.; Di Raimondo, D.; Di Sciacca, R.; Arnao, V.; Licata, G. Immuno-inflammatory predictors of stroke at follow-up in patients with chronic non-valvular atrial fibrillation (NVAF). Clin. Sci. 2009, 116, 781–789. [Google Scholar] [CrossRef] [Green Version]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Hak, L.; Mysliwska, J.; Wieckiewicz, J.; Szyndler, K.; Siebert, J.; Rogowski, J. Interleukin-2 as a predictor of early postoperative atrial fibrillation after cardiopulmonary bypass graft (CABG). J. Interferon Cytokine Res. 2009, 29, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Guillot-Sestier, M.-V.; Doty, K.R.; Gate, D.; Rodriguez, J., Jr.; Leung, B.P.; Rezai-Zadeh, K.; Town, T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron 2015, 85, 534–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alegret, J.M.; Aragonès, G.; Elosua, R.; Beltrán-Debón, R.; Hernández-Aguilera, A.; Romero-Menor, C.; Camps, J.; Joven, J. The relevance of the association between inflammation and atrial fibrillation. Eur. J. Clin. Investig. 2013, 43, 324–331. [Google Scholar] [CrossRef]
- Lee, W.-J.; Liao, Y.-C.; Wang, Y.-F.; Lin, I.-F.; Wang, S.-J.; Fuh, J.-L. Plasma MCP-1 and cognitive decline in patients with Alzheimer’s disease and mild cognitive impairment: A two-year follow-up study. Sci. Rep. 2018, 8, 1280. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, M.; Arena, R. Endothelial dysfunction and pathophysiological correlates in atrial fibrillation. Heart 2009, 95, 102–106. [Google Scholar] [CrossRef]
- Bunch, T.J.; Galenko, O.; Graves, K.G.; Jacobs, V.; May, H.T. Atrial Fibrillation and Dementia: Exploring the Association, Defining Risks and Improving Outcomes. Arrhythm. Electrophysiol. Rev. 2019, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Lip, G.Y.; Apostolakis, S. Inflammation in atrial fibrillation. J. Am. Coll. Cardiol. 2012, 60, 2263–2270. [Google Scholar] [CrossRef] [Green Version]
- Siasos, G.; Mazaris, S.; Zisimos, K.; Oikonomou, E.; Kokkou, E.; Konsola, T.; Mourouzis, K.; Vavuranakis, M.; Zografos, T.; Zaromitidou, M. The impact of atrial fibrillation on endothelial dysfunction. J. Am. Coll. Cardiol. 2015, 65, A477. [Google Scholar] [CrossRef]
- Weymann, A.; Sabashnikov, A.; Ali-Hasan-Al-Saegh, S.; Popov, A.-F.; Mirhosseini, S.J.; Baker, W.L.; Lotfaliani, M.; Liu, T.; Dehghan, H.; Yavuz, S. Predictive role of coagulation, fibrinolytic, and endothelial markers in patients with atrial fibrillation, stroke, and thromboembolism: A meta-analysis, meta-regression, and systematic review. Med. Sci. Monit. Basic Res. 2017, 23, 97. [Google Scholar]
- Kelleher, R.J.; Soiza, R.L. Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’sa vascular disorder? Am. J. Cardiovasc. Dis. 2013, 3, 197. [Google Scholar]
- Herrera, M.D.; Mingorance, C.; Rodríguez-Rodríguez, R.; De Sotomayor, M.A. Endothelial dysfunction and aging: An update. Ageing Res. Rev. 2010, 9, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Montenero, A.S.; Mollichelli, N.; Zumbo, F.; Antonelli, A.; Dolci, A.; Barberis, M.; Sirolla, C.; Staine, T.; Fiocca, L.; Bruno, N. Helicobacter pylori and atrial fibrillation: A possible pathogenic link. Heart 2005, 91, 960–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kountouras, J.; Boziki, M.; Gavalas, E.; Zavos, C.; Deretzi, G.; Grigoriadis, N.; Tsolaki, M.; Chatzopoulos, D.; Katsinelos, P.; Tzilves, D. Increased cerebrospinal fluid Helicobacter pylori antibody in Alzheimer’s disease. Int. J. Neurosci. 2009, 119, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-S.; Yang, T.-Y.; Shen, W.-C.; Lin, C.-L.; Lin, M.-C.; Kao, C.-H. Association between Helicobacter pylori infection and dementia. J. Clin. Neurosci. 2014, 21, 1355–1358. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, M.A.; Beydoun, H.A.; Shroff, M.R.; Kitner-Triolo, M.H.; Zonderman, A.B. Helicobacter pylori seropositivity and cognitive performance among US adults: Evidence from a large national survey. Psychosom. Med. 2013, 75, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, A.G.; Zagari, R.M.; De Musis, C.; Romano, L.; Loguercio, C.; Romano, M. Helicobacter pylori and extragastric diseases: A review. World J. Gastroenterol. 2018, 24, 3204. [Google Scholar] [CrossRef]
- Chiang, C.-H.; Huang, C.-C.; Chan, W.-L.; Huang, P.-H.; Chen, Y.-C.; Chen, T.-J.; Lin, S.-J.; Chen, J.-W.; Leu, H.-B. Herpes simplex virus infection and risk of atrial fibrillation: A nationwide study. Int. J. Cardiol. 2013, 164, 201–204. [Google Scholar] [CrossRef]
- Lövheim, H.; Gilthorpe, J.; Johansson, A.; Eriksson, S.; Hallmans, G.; Elgh, F. Herpes simplex infection and the risk of Alzheimer’s disease: A nested case-control study. Alzheimers Dement. 2015, 11, 587–592. [Google Scholar] [CrossRef]
- Lövheim, H.; Gilthorpe, J.; Adolfsson, R.; Nilsson, L.-G.; Elgh, F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 593–599. [Google Scholar] [CrossRef]
- Wozniak, M.; Mee, A.; Itzhaki, R. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef]
- Itzhaki, R.F. Corroboration of a major role for herpes simplex virus type 1 in Alzheimer’s disease. Front Aging Neurosci. 2018, 10, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krijthe, B.P.; Heeringa, J.; Hofman, A.; Franco, O.H.; Stricker, B.H. Non-steroidal anti-inflammatory drugs and the risk of atrial fibrillation: A population-based follow-up study. BMJ Open 2014, 4, e004059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Christiansen, C.F.; Mehnert, F.; Rothman, K.J.; Sørensen, H.T. Non-steroidal anti-inflammatory drug use and risk of atrial fibrillation or flutter: Population based case-control study. BMJ 2011, 343, d3450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslam, A.K.; Vasavada, B.C.; Sacchi, T.J.; Khan, I.A. Atrial fibrillation associated with systemic lupus erythematosus and use of methylprednisolone. Am. J. Ther. 2001, 8, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Whelton, A. Renal aspects of treatment with conventional nonsteroidal anti-inflammatory drugs versus cyclooxygenase-2–specific inhibitors. Am. J. Med. 2001, 110, 33–42. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, J.; Zhang, F. NSAID Exposure and Risk of Alzheimer’s Disease: An Updated Meta-Analysis from Cohort Studies. Front. Aging Neurosci. 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kaida-Yip, F.; Zabel, M. NSAID Use and the Prevention of Alzheimer’s Disease: A Meta-Analysis (P6. 184). Neurology 2018, 90, 6-184. [Google Scholar]
- Shiroshita-Takeshita, A.; Brundel, B.J.; Lavoie, J.; Nattel, S. Prednisone prevents atrial fibrillation promotion by atrial tachycardia remodeling in dogs. Cardiovasc. Res. 2006, 69, 865–875. [Google Scholar] [CrossRef] [Green Version]
- Ho, K.M.; Tan, J.A. Benefits and risks of corticosteroid prophylaxis in adult cardiac surgery: A dose-response meta-analysis. Circulation 2009, 119, 1853–1866. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Wang, J.; Yiu, D.; Liu, K. The Efficacy of Glucocorticoids for the Prevention of Atrial Fibrillation, or Length of Intensive Care Unite or Hospital Stay After Cardiac Surgery: A Meta-Analysis. Cardiovasc. Ther. 2014, 32, 89–96. [Google Scholar] [CrossRef]
- Koyama, T.; Tada, H.; Sekiguchi, Y.; Arimoto, T.; Yamasaki, H.; Kuroki, K.; Machino, T.; Tajiri, K.; Zhu, X.D.; Kanemoto-Igarashi, M. Prevention of atrial fibrillation recurrence with corticosteroids after radiofrequency catheter ablation: A randomized controlled trial. J. Am. Coll. Cardiol. 2010, 56, 1463–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansen, C.F.; Christensen, S.; Mehnert, F.; Cummings, S.R.; Chapurlat, R.D.; Sørensen, H.T. Glucocorticoid use and risk of atrial fibrillation or flutter: A population-based, case-control study. Arch. Intern. Med. 2009, 169, 1677–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Hooft, C.S.; Heeringa, J.; Brusselle, G.G.; Hofman, A.; Witteman, J.C.; Kingma, J.H.; Sturkenboom, M.C.; Stricker, B.H.C. Corticosteroids and the risk of atrial fibrillation. Arch. Intern. Med. 2006, 166, 1016–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; MacDonald, T.M.; Walker, B.R. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann. Intern. Med. 2004, 141, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Beeri, M.S.; Schmeidler, J.; Lesser, G.T.; Maroukian, M.; West, R.; Leung, S.; Wysocki, M.; Perl, D.P.; Purohit, D.P.; Haroutunian, V. Corticosteroids, but not NSAIDs, are associated with less Alzheimer neuropathology. Neurobiol. Aging 2012, 33, 1258–1264. [Google Scholar] [CrossRef] [Green Version]
- Aisen, P.S.; Davis, K.; Berg, J.; Schafer, K.; Campbell, K.; Thomas, R.; Weiner, M.; Farlow, M.; Sano, M.; Grundman, M. A randomized controlled trial of prednisone in Alzheimer’s disease. Neurology 2000, 54, 588. [Google Scholar] [CrossRef]
- Libro, R.; Bramanti, P.; Mazzon, E. Endogenous glucocorticoids: Role in the etiopathogenesis of Alzheimer’s disease. Neuro Endocrinol. Lett. 2017, 38, 1–12. [Google Scholar]
- Mandal, K.; Torsney, E.; Poloniecki, J.; Camm, A.J.; Xu, Q.; Jahangiri, M. Association of High Intracellular, But Not Serum, Heat Shock Protein 70 With Postoperative Atrial Fibrillation. Ann. Thorac. Cardiovasc. Surg. 2005, 79, 865–871. [Google Scholar] [CrossRef]
- Hu, Y.-F.; Yeh, H.-I.; Tsao, H.-M.; Tai, C.-T.; Lin, Y.-J.; Chang, S.-L.; Lo, L.-W.; Tuan, T.-C.; Suenari, K.; Li, C.-H. Electrophysiological correlation and prognostic impact of heat shock protein 27 in atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2012, 5, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.-Q.; Shen, W.; Chen, J.; Wang, B.-R.; Zhong, L.-L.; Zhu, Y.-W.; Zhu, H.-Q.; Zhang, Q.-Q.; Zhang, Y.-D.; Xu, J. Anti-TNF-α reduces amyloid plaques and tau phosphorylation and induces CD11c-positive dendritic-like cell in the APP/PS1 transgenic mouse brains. Brain Res. 2011, 1368, 239–247. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and tau effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tweedie, D.; Ferguson, R.A.; Fishman, K.; Frankola, K.A.; Van Praag, H.; Holloway, H.W.; Luo, W.; Li, Y.; Caracciolo, L.; Russo, I. Tumor necrosis factor-α synthesis inhibitor 3, 6′-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J. Neuroinflammation 2012, 9, 106. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-C.; Hung, C.-Y.; Li, C.-H.; Liao, Y.-C.; Huang, J.-L.; Lin, C.-H.; Wu, T.-J. Angiotensin-receptor blocker, angiotensin-converting enzyme inhibitor, and risks of atrial fibrillation: A nationwide cohort study. Medicine 2016, 95, e3721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, Y.; Liu, Q.; Shan, Q. Effects of renin–angiotensin–aldosterone system inhibitors on mortality, hospitalization, and diastolic function in patients with HFpEF. Herz 2016, 41, 76–86. [Google Scholar] [CrossRef]
- Yasar, S.; Xia, J.; Yao, W.; Furberg, C.D.; Xue, Q.-L.; Mercado, C.I.; Fitzpatrick, A.L.; Fried, L.P.; Kawas, C.H.; Sink, K.M. Antihypertensive drugs decrease risk of Alzheimer disease: Ginkgo Evaluation of Memory Study. Neurology 2013, 81, 896–903. [Google Scholar] [CrossRef] [Green Version]
- Barthold, D.; Joyce, G.; Wharton, W.; Kehoe, P.; Zissimopoulos, J. The association of multiple anti-hypertensive medication classes with Alzheimer’s disease incidence across sex, race, and ethnicity. PLoS ONE 2018, 13, e0206705. [Google Scholar] [CrossRef]
- Feng, B.; Xu, L.; Wang, H.; Yan, X.; Xue, J.; Liu, F.; Hu, J.-F. Atorvastatin exerts its anti-atherosclerotic effects by targeting the receptor for advanced glycation end products. Biochim. Biophys. Acta 2011, 1812, 1130–1137. [Google Scholar] [CrossRef]
- An, L.; An, S.; Jia, Z.; Wang, H.; Yang, Z.; Xu, C.; Teng, X.; Wang, J.; Liu, X.; Cao, Q. Atorvastatin improves left ventricular remodeling and cardiac function in rats with congestive heart failure by inhibiting RhoA/Rho kinase-mediated endothelial nitric oxide synthase. Exp. Ther. Med. 2019, 17, 960–966. [Google Scholar] [CrossRef] [Green Version]
- Warita, S.; Kawasaki, M.; Tanaka, R.; Ono, K.; Kojima, T.; Hirose, T.; Iwama, M.; Watanabe, T.; Nishigaki, K.; Takemura, G. Effects of pitavastatin on cardiac structure and function and on prevention of atrial fibrillation in elderly hypertensive patients. Circ. J. 2012, 76, 2755–2762. [Google Scholar] [CrossRef] [Green Version]
- Shiroshita-Takeshita, A.; Schram, G.; Lavoie, J.; Nattel, S. Effect of simvastatin and antioxidant vitamins on atrial fibrillation promotion by atrial-tachycardia remodeling in dogs. Circulation 2004, 110, 2313–2319. [Google Scholar] [CrossRef] [Green Version]
- Maesen, B.; Nijs, J.; Maessen, J.; Allessie, M.; Schotten, U. Post-operative atrial fibrillation: A maze of mechanisms. Europace 2011, 14, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Liakopoulos, O.J.; Choi, Y.-H.; Kuhn, E.W.; Wittwer, T.; Borys, M.; Madershahian, N.; Wassmer, G.; Wahlers, T. Statins for prevention of atrial fibrillation after cardiac surgery: A systematic literature review. J. Thorac. Cardiovasc. Surg. 2009, 138, 678–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, C.N.; Greve, A.M.; Abdulla, J.; Køber, L.; Gislason, G.H.; Wachtell, K. The preventive effect of statin therapy on new-onset and recurrent atrial fibrillation in patients not undergoing invasive cardiac interventions: A systematic review and meta-analysis. Int. J. Cardiol. 2013, 167, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Sinyavskaya, L.; Gauthier, S.; Renoux, C.; Dell’Aniello, S.; Suissa, S.; Brassard, P. Comparative effect of statins on the risk of incident Alzheimer disease. Neurology 2018, 90, e179–e187. [Google Scholar] [CrossRef]
- Chu, C.-S.; Tseng, P.-T.; Stubbs, B.; Chen, T.-Y.; Tang, C.-H.; Li, D.-J.; Yang, W.-C.; Chen, Y.-W.; Wu, C.-K.; Veronese, N. Use of statins and the risk of dementia and mild cognitive impairment: A systematic review and meta-analysis. Sci. Rep. 2018, 8, 5804. [Google Scholar] [CrossRef]
- Deftereos, S.; Giannopoulos, G.; Kossyvakis, C.; Efremidis, M.; Panagopoulou, V.; Kaoukis, A.; Raisakis, K.; Bouras, G.; Angelidis, C.; Theodorakis, A. Colchicine for prevention of early atrial fibrillation recurrence after pulmonary vein isolation: A randomized controlled study. J. Am. Coll. Cardiol. 2012, 60, 1790–1796. [Google Scholar] [CrossRef] [Green Version]
- Imazio, M.; Brucato, A.; Ferrazzi, P.; Pullara, A.; Adler, Y.; Barosi, A.; Caforio, A.L.; Cemin, R.; Chirillo, F.; Comoglio, C. Colchicine for prevention of postpericardiotomy syndrome and postoperative atrial fibrillation: The COPPS-2 randomized clinical trial. JAMA 2014, 312, 1016–1023. [Google Scholar] [CrossRef]
- Aisen, P.S.; Marin, D.B.; Brickman, A.M.; Santoro, J.; Fusco, M. Pilot tolerability studies of hydroxychloroquine and colchicine in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2001, 15, 96–101. [Google Scholar] [CrossRef]
- Kumar, A.; Seghal, N.; Naidu, P.S.; Padi, S.S.; Goyal, R. Colchicines-induced neurotoxicity as an animal model of sporadic dementia of Alzheimer’s type. Pharmacol. Rep. 2007, 59, 274. [Google Scholar]
- Wu, J.H.; Lemaitre, R.N.; King, I.B.; Song, X.; Sacks, F.M.; Rimm, E.B.; Heckbert, S.R.; Siscovick, D.S.; Mozaffarian, D. Association of Plasma Phospholipid Long-Chain Omega-3 Fatty Acids with Incident Atrial Fibrillation in Older AdultsClinical Perspective: The Cardiovascular Health Study. Circulation 2012, 125, 1084–1093. [Google Scholar] [CrossRef]
- Kumar, S.; Sutherland, F.; Morton, J.B.; Lee, G.; Morgan, J.; Wong, J.; Eccleston, D.E.; Voukelatos, J.; Garg, M.L.; Sparks, P.B. Long-term omega-3 polyunsaturated fatty acid supplementation reduces the recurrence of persistent atrial fibrillation after electrical cardioversion. Heart Rhythm 2012, 9, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Nigam, A.; Talajic, M.; Roy, D.; Nattel, S.; Lambert, J.; Nozza, A.; Jones, P.; Ramprasath, V.R.; O’Hara, G.; Kopecky, S. Fish oil for the reduction of atrial fibrillation recurrence, inflammation, and oxidative stress. J. Am. Coll. Cardiol. 2014, 64, 1441–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Zhen, Y.; Tao, A.; Bao, Z.; Zhang, G. Polyunsaturated fatty acids for the prevention of atrial fibrillation after cardiac surgery: An updated meta-analysis of randomized controlled trials. J. Cardiol. 2014, 63, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shchepinov, M.; Mattson, M.; Bennett, B. A new treatment Paradigm for Neurodegeneration: Peroxidation-resistant polyunsaturated fatty acids (D-PUFA) lower brain amyloid beta and oxidation markers, and reverse cognition impairment in vivo (P4. 090). Neurology 2017, 88, 4-090. [Google Scholar]
- Phillips, M.; Childs, C.; Calder, P.; Rogers, P. No effect of omega-3 fatty acid supplementation on cognition and mood in individuals with cognitive impairment and probable Alzheimer’s disease: A randomised controlled trial. Int. J. Mol. Sci. 2015, 16, 24600–24613. [Google Scholar] [CrossRef]
- Wagner, K.M.; McReynolds, C.B.; Schmidt, W.K.; Hammock, B.D. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol. Ther. 2017, 180, 62–76. [Google Scholar] [CrossRef]
- Sirish, P.; Li, N.; Timofeyev, V.; Zhang, X.-D.; Wang, L.; Yang, J.; Lee, K.S.S.; Bettaieb, A.; Ma, S.M.; Lee, J.H. Molecular mechanisms and new treatment paradigm for atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2016, 9, e003721. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Liu, J.-Y.; Timofeyev, V.; Qiu, H.; Hwang, S.H.; Tuteja, D.; Lu, L.; Yang, J.; Mochida, H.; Low, R. Beneficial effects of soluble epoxide hydrolase inhibitors in myocardial infarction model: Insight gained using metabolomic approaches. J. Mol. Cell. Cardiol. 2009, 47, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Li, N.; Liu, J.Y.; Harris, T.R.; Hammock, B.D.; Chiamvimonvat, N. Soluble epoxide hydrolase inhibitors and heart failure. Cardiovasc. Ther. 2011, 29, 99–111. [Google Scholar] [CrossRef]
- Wutzler, A.; Kestler, C.; Perrot, A.; Loehr, L.; Huemer, M.; Parwani, A.S.; Attanasio, P.; Özcelik, C.; Schunck, W.-H.; Gollasch, M. Variations in the human soluble epoxide hydrolase gene and recurrence of atrial fibrillation after catheter ablation. Int. J. Cardiol. 2013, 168, 3647–3651. [Google Scholar] [CrossRef]
- Sarkar, P.; Narayanan, J.; Harder, D.R. Differential effect of amyloid beta on the cytochrome P450 epoxygenase activity in rat brain. Neuroscience 2011, 194, 241–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, P.; Zaja, I.; Bienengraeber, M.; Rarick, K.R.; Terashvili, M.; Canfield, S.; Falck, J.R.; Harder, D.R. Epoxyeicosatrienoic acids pretreatment improves amyloid β-induced mitochondrial dysfunction in cultured rat hippocampal astrocytes. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H475–H484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-T.; Lee, K.-I.; Chen, C.-H.; Lee, T.-S. Genetic deletion of soluble epoxide hydrolase delays the progression of Alzheimer’s disease. J. Neuroinflammation 2019, 16, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griñán-Ferré, C.; Codony, S.; Pujol, E.; Yang, J.; Leiva, R.; Escolano, C.; Puigoriol-Illamola, D.; Companys-Alemany, J.; Corpas, R.; Sanfeliu, C. Pharmacological inhibition of soluble epoxide hydrolase as a new therapy for Alzheimer’s Disease. bioRxiv 2019, 605055. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, V.; Woller, S.C.; Stevens, S.; May, H.T.; Bair, T.L.; Anderson, J.L.; Crandall, B.G.; Day, J.D.; Johanning, K.; Long, Y. Time outside of therapeutic range in atrial fibrillation patients is associated with long-term risk of dementia. Heart Rhythm 2014, 11, 2206–2213. [Google Scholar] [CrossRef]
- Jacobs, V.; May, H.T.; Bair, T.L.; Crandall, B.G.; Cutler, M.J.; Day, J.D.; Mallender, C.; Osborn, J.S.; Stevens, S.M.; Weiss, J.P. Long-term population-based cerebral ischemic event and cognitive outcomes of direct oral anticoagulants compared with warfarin among long-term anticoagulated patients for atrial fibrillation. J. Am. Coll. Cardiol. 2016, 118, 210–214. [Google Scholar] [CrossRef]
- Bunch, T.J.; Crandall, B.G.; Weiss, J.P.; May, H.T.; Bair, T.L.; Osborn, J.S.; Anderson, J.L.; Muhlestein, J.B.; Horne, B.D.; Lappe, D.L. Patients treated with catheter ablation for atrial fibrillation have long-term rates of death, stroke, and dementia similar to patients without atrial fibrillation. J. Cardiovasc. Electrophysiol. 2011, 22, 839–845. [Google Scholar] [CrossRef]
- Bordier, P.; Lanusse, S.; Garrigue, S.; Reynard, C.; Robert, F.; Gencel, L.; Lafitte, A. Causes of syncope in patients with Alzheimer’s disease treated with donepezil. Drugs Aging 2005, 22, 687–694. [Google Scholar] [CrossRef]
- Malone, D.M.; Lindesay, J. Cholinesterase inhibitors and cardiovascular disease: A survey of old age psychiatrists’ practice. Age Ageing 2007, 36, 331–333. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-T.; Wu, P.-H.; Chen, C.-S.; Yang, Y.-H.; Yang, Y.-H. Association between acetylcholinesterase inhibitors and risk of stroke in patients with dementia. Sci. Rep. 2016, 6, 29266. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Parameter | Atrial Fibrillation | Alzheimer’s Disease |
|---|---|---|
| IL-1β | In the atrium of patients with AF, in comparison to controls, an increase in pro-inflammatory macrophages was observed, which was associated with a higher secretion of IL-1β [37]. It was also related to an up-regulation of IL-1β mRNA in the atrium of patients with persistent AF in comparison to those with paroxysmal AF and the control group [43]. | IL-1β demonstrated a key function in the deposition of βAP plaques [44]. Higher levels of IL-1β were found in the serum samples of both patients with AD and mild cognition impairment (MCI), leading to the consideration that this cytokine is produced in the beginning of the disease and remains elevated after the establishment of AD [23,45]. |
| IL-6 | IL-6 was related to the development of AF after coronary artery bypass grafting (CABG) [38]. IL-6 was also associated with the recurrence of AF after ablation therapy [46]. | IL-6 was associated with βAP plaques in the hippocampus and cortex in the AD brain [47] and to abnormally hyperphosphorylated tau protein [48]. Higher levels of IL-6 were found in the serum of AD patients compared to those with MCI and the control group [40]. |
| TNF | Higher levels of TNF were detected in patients with persistent and permanent AF in comparison to those with paroxysmal AF [39]. The TNF values found in the plasma of patients with chronic non-valvular AF were also considered as predictors for the development of ischemic strokes [49]. | TNF was associated with an increase in βAP production in an experimental study [41]. In a meta-analysis, an up-regulation of TNF in the blood and CSF samples of patients with AD was reported, especially in those in severe stages of the disease [50]. |
| IL-10 | Increased levels of IL-10 were found in peripheral blood samples of AF patients compared to the controls, and higher concentrations were detected in patients with persistent and permanent AF compared to those with paroxysmal AF [39]. Higher serum levels of IL-10 were also associated with the development of AF after CABG [51]. | IL-10 was related to the accumulation of βAP in an animal model [52]. In a clinical study, AD patients exhibited higher serum levels of IL-10 than the controls, suggesting that peripheral levels of IL-10 may be related to AD pathogenesis [42]. |
| MCP-1 | Higher levels of monocyte chemo-attractant protein (MCP)-1 were found in venous blood samples from patients with AF compared to the controls [39]. Elevated levels of this marker were also found in the plasma of patients with paroxysmal and permanent AF in relation to individuals with sinus rhythm [53]. | Higher levels of MCP-1 were found in AD patients compared with MCI patients and controls, and the highest levels were assessed in severe AD patients. Thus, higher plasmatic levels of MCP-1 were associated with a greater severity of AD [54]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, G.L.; Duarte, R.C.F.; Mukhamedyarov, M.A.; Palotás, A.; Ferreira, C.N.; Reis, H.J. Inflammatory and Infectious Processes Serve as Links between Atrial Fibrillation and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3226. https://doi.org/10.3390/ijms21093226
Martins GL, Duarte RCF, Mukhamedyarov MA, Palotás A, Ferreira CN, Reis HJ. Inflammatory and Infectious Processes Serve as Links between Atrial Fibrillation and Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(9):3226. https://doi.org/10.3390/ijms21093226
Chicago/Turabian StyleMartins, Gabriela Lopes, Rita Carolina Figueiredo Duarte, Marat Alexandrovich Mukhamedyarov, András Palotás, Cláudia Natália Ferreira, and Helton José Reis. 2020. "Inflammatory and Infectious Processes Serve as Links between Atrial Fibrillation and Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 9: 3226. https://doi.org/10.3390/ijms21093226