New Omics—Derived Perspectives on Retinal Dystrophies: Could Ion Channels-Encoding or Related Genes Act as Modifier of Pathological Phenotype?

 , ,
, ,  and
and
Abstract
:1. Introduction
The Physiological Role of Main Ocular Ion Channel Types and Their Association to Eye-Related Diseases
2. Results
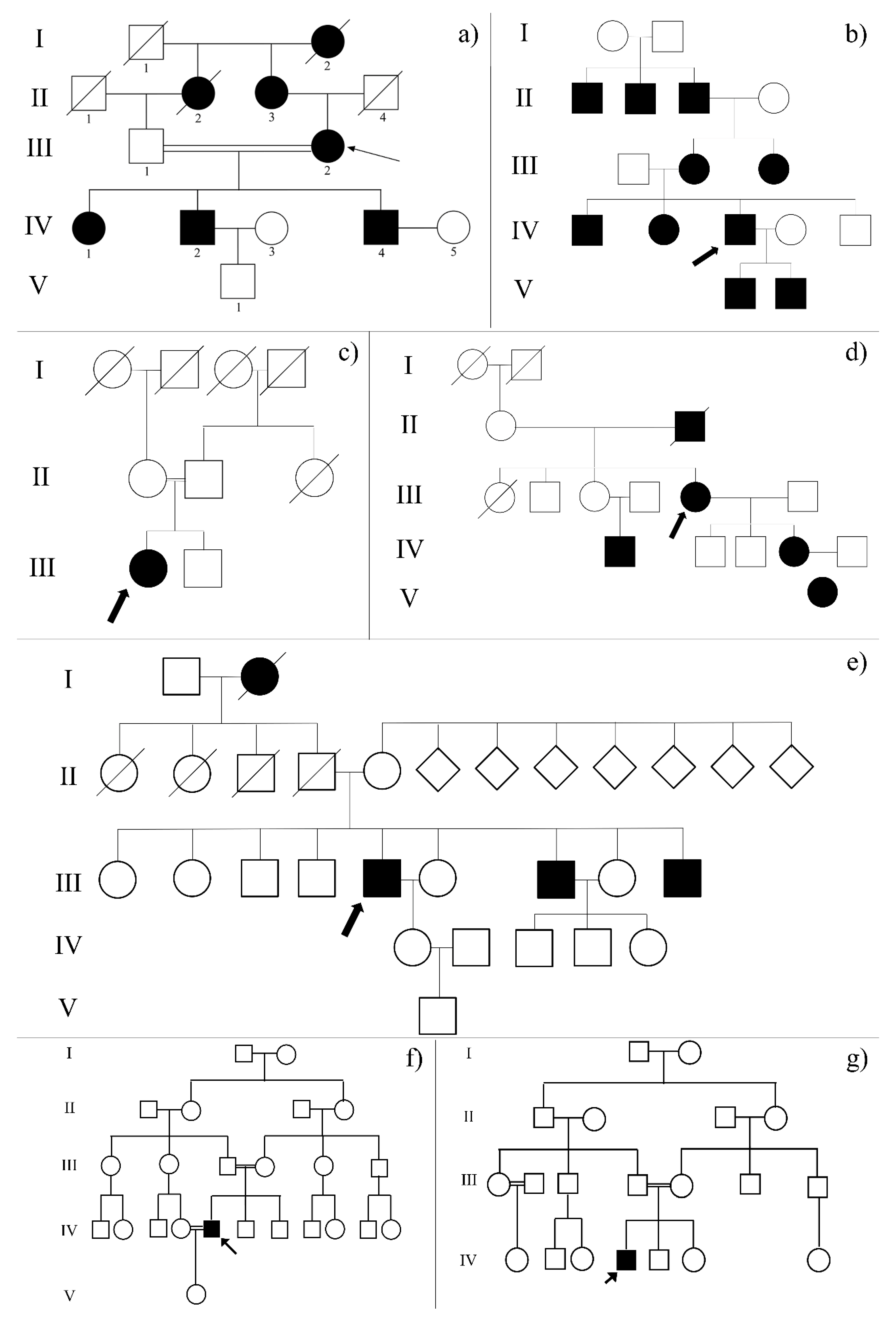
2.1. Clinical Examination of Family Probands Highlighted the Possible Impairment of Retinal Neurotransmission
2.2. Whole Exome Sequencing Data Analysis Revealed More than One Known Causative Variant of Retinal Dystrophies in Each Proband
2.3. The Complex Genotype-Phenotype Association Suggested the Possible Involvement of Modifier Genes Encoding Ion Channels
2.4. Biochemical Analyses Highlighted Possible Altered Chemical-Physical Features in Mutated Channels
2.5. 3D Structure Analysis of Mutated Channels Showed the Deletion or the Addition of Ion-Binding Sites
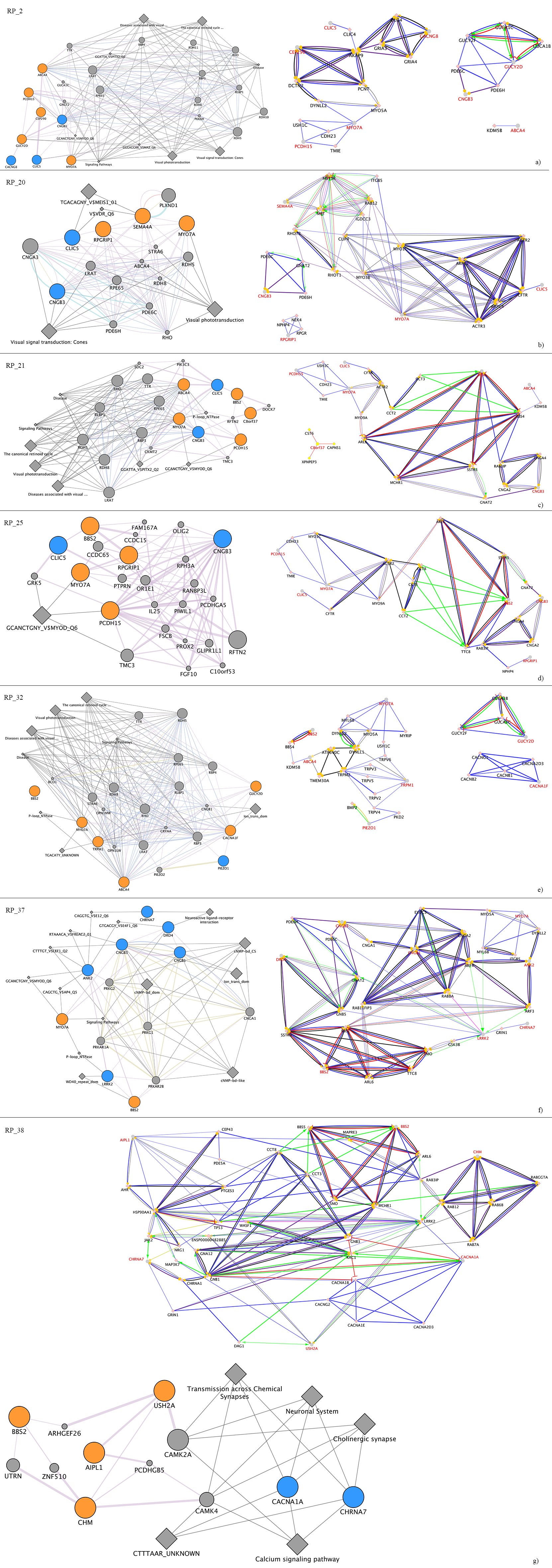
2.6. Pathways Analysis of Ion Channel-Related Mutated Genes Suggested a Complex Regulation of Synaptic Transmission Associated to Light Stimuli
2.7. Ion Channel-Related Mutated Genes and Known Causative Genes of Retinal Dystrophies Suggested a Complex Regulation Network of Interactors
3. Discussion
4. Materials and Methods
4.1. Clinical Data and Sample Collection
4.2. Whole Exome Sequencing and Data Analysis
4.3. Variant Filtering and Gene Prioritization
4.4. Variant Validation
4.5. Proteomic in-Silico Analyses
4.6. Pathway and Subpathway Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| IRDs | Inherited Retinal Dystrophies |
| RPE | Retinal Pigment Epithelium |
| RGCs | Retinal Ganglion Cells |
| CFTR | Cystic Fibrosis Transmembrane Conductance Regulator |
| LCA | Leber Congenital Amaurosis |
| RP | Retinitis Pigmentosa |
| ERG | Electroretinogram |
| VEP | Visual Evoked Potential |
| HGMD | Human Gene Mutation Database |
| WES | Whole Exome Sequencing |
| GO | Gene Ontology |
| PTC | Premature Termination Codon |
| AMPAR | AMPA Receptors |
| BCVA | Best-Corrected Visual Acuity |
| SD-OCT | Spectral-Domain Optical Coherence Tomography |
| ISCEV | International Society for Clinical Electrophysiology of Vision |
| PCR | Polymerase Chain Reaction |
| RMSD | Root Mean Square Deviation |
References
- Gonzalez-Duarte, R.; de Castro-Miro, M.; Tuson, M.; Ramirez-Castaneda, V.; Gils, R.V.; Marfany, G. Scaling New Heights in the Genetic Diagnosis of Inherited Retinal Dystrophies. Adv. Exp. Med. Biol. 2019, 1185, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.G.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.R.; Benson, M.D.; MacDonald, I.M.; Innes, A.M. A diagnostic approach to syndromic retinal dystrophies with intellectual disability. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 538–570. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, R.; Donato, L.; Venza, I.; Scimone, C.; Aragona, P.; Sidoti, A. Possible protective role of the ABCA4 gene c.1268A>G missense variant in Stargardt disease and syndromic retinitis pigmentosa in a Sicilian family: Preliminary data. Int. J. Mol. Med. 2017, 39, 1011–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scimone, C.; Donato, L.; Esposito, T.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. A novel RLBP1 gene geographical area-related mutation present in a young patient with retinitis punctata albescens. Hum. Genom. 2017, 11, 18. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Nicocia, G.; Denaro, L.; Robledo, R.; Sidoti, A.; D’Angelo, R. GLO1 gene polymorphisms and their association with retinitis pigmentosa: A case-control study in a Sicilian population. Mol. Biol. Rep. 2018. [Google Scholar] [CrossRef]
- Donato, L.; Bramanti, P.; Scimone, C.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. miRNAexpression profile of retinal pigment epithelial cells under oxidative stress conditions. FEBS Open Bio. 2018, 8, 219–233. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; D’Angelo, R.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; Scimone, C. Effects of A2E-Induced Oxidative Stress on Retinal Epithelial Cells: New Insights on Differential Gene Response and Retinal Dystrophies. Antioxidants 2020, 9, 307. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; Scimone, C.; Alibrandi, S.; Nicocia, G.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Discovery of GLO1 New Related Genes and Pathways by RNA-Seq on A2E-Stressed Retinal Epithelial Cells Could Improve Knowledge on Retinitis Pigmentosa. Antioxidants 2020, 9, 416. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Pitruzzella, A.; Scalia, F.; D’Angelo, R.; Sidoti, A. Possible A2E Mutagenic Effects on RPE Mitochondrial DNA from Innovative RNA-Seq Bioinformatics Pipeline. Antioxidants 2020, 9, 1158. [Google Scholar] [CrossRef] [PubMed]
- Donato, L.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Sidoti, A.; D’Angelo, R. Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa. Antioxidants 2020, 9, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scimone, C.; Alibrandi, S.; Scalinci, S.Z.; Trovato Battagliola, E.; D’Angelo, R.; Sidoti, A.; Donato, L. Expression of Pro-Angiogenic Markers Is Enhanced by Blue Light in Human RPE Cells. Antioxidants 2020, 9, 1154. [Google Scholar] [CrossRef] [PubMed]
- Hohman, T.C. Hereditary Retinal Dystrophy. Handb. Exp. Pharmacol. 2017, 242, 337–367. [Google Scholar] [CrossRef] [PubMed]
- Branham, K.; Schlegel, D.; Fahim, A.T.; Jayasundera, K.T. Genetic testing for inherited retinal degenerations: Triumphs and tribulations. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 571–577. [Google Scholar] [CrossRef]
- Giblin, J.P.; Comes, N.; Strauss, O.; Gasull, X. Ion Channels in the Eye: Involvement in Ocular Pathologies. Adv. Protein Chem. Struct. Biol. 2016, 104, 157–231. [Google Scholar] [CrossRef]
- Reichhart, N.; Strauss, O. Ion channels and transporters of the retinal pigment epithelium. Exp. Eye Res. 2014, 126, 27–37. [Google Scholar] [CrossRef]
- Zhong, Y.S.; Wang, J.; Liu, W.M.; Zhu, Y.H. Potassium ion channels in retinal ganglion cells (review). Mol. Med. Rep. 2013, 8, 311–319. [Google Scholar] [CrossRef]
- Reigada, D.; Mitchell, C.H. Release of ATP from retinal pigment epithelial cells involves both CFTR and vesicular transport. Am. J. Physiol. Cell Physiol. 2005, 288, C132–C140. [Google Scholar] [CrossRef]
- Fesenko, E.E.; Kolesnikov, S.S.; Lyubarsky, A.L. Induction by cyclic GMP of cationic conductance in plasma membrane of retinal rod outer segment. Nature 1985, 313, 310–313. [Google Scholar] [CrossRef]
- Michalakis, S.; Becirovic, E.; Biel, M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int. J. Mol. Sci. 2018, 19, 749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Pang, S.; Wang, J.; FitzMaurice, B.; Pang, J.; Chang, B. Photoreceptor degeneration in a new Cacna1f mutant mouse model. Exp. Eye Res. 2019, 179, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.P.; Zhao, J.W.; Yang, X.L. Expression of voltage-dependent calcium channel subunits in the rat retina. Neurosci. Lett. 2002, 329, 297–300. [Google Scholar] [CrossRef]
- Kersten, F.F.; van Wijk, E.; van Reeuwijk, J.; van der Zwaag, B.; Marker, T.; Peters, T.A.; Katsanis, N.; Wolfrum, U.; Keunen, J.E.; Roepman, R.; et al. Association of whirlin with Cav1.3 (alpha1D) channels in photoreceptors, defining a novel member of the usher protein network. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2338–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsokolas, G.; Almuhtaseb, H.; Griffiths, H.; Shawkat, F.; Pengelly, R.J.; Sarah, E.; Lotery, A. Long term follow-up of a family with GUCY2D dominant cone dystrophy. Int. J. Ophthalmol. 2018, 11, 1945–1950. [Google Scholar] [CrossRef]
- Kabra, M.; Pattnaik, B.R. Sensing through Non-Sensing Ocular Ion Channels. Int. J. Mol. Sci. 2020, 21, 6925. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Zha, X.M. Acid-sensing ion channels: Trafficking and synaptic function. Mol. Brain 2013, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Tanner, M.R.; Beeton, C. Differences in ion channel phenotype and function between humans and animal models. Front. Biosci. 2018, 23, 43–64. [Google Scholar] [CrossRef] [Green Version]
- Ohya, S.; Kito, H.; Hatano, N.; Muraki, K. Recent advances in therapeutic strategies that focus on the regulation of ion channel expression. Pharmacol. Ther. 2016, 160, 11–43. [Google Scholar] [CrossRef] [PubMed]
- Di Resta, C.; Spiga, I.; Presi, S.; Merella, S.; Pipitone, G.B.; Manitto, M.P.; Querques, G.; Parodi, M.B.; Ferrari, M.; Carrera, P. Integration of multigene panels for the diagnosis of hereditary retinal disorders using Next Generation Sequencing and bioinformatics approaches. eJIFCC 2018, 29, 15–25. [Google Scholar] [PubMed]
- Tetreault, M.L.; Horrigan, D.M.; Kim, J.A.; Zimmerman, A.L. Retinoids restore normal cyclic nucleotide sensitivity of mutant ion channels associated with cone dystrophy. Mol. Vis. 2006, 12, 1699–1705. [Google Scholar] [PubMed]
- Koenekoop, R.K. Revisiting Congenital Stationary Night Blindness in the Molecular Era. JAMA Ophthalmol. 2018, 136, 398–399. [Google Scholar] [CrossRef]
- Abdelkader, E.; Brandau, O.; Bergmann, C.; AlSalamah, N.; Nowilaty, S.; Schatz, P. Novel causative variants in patients with achromatopsia. Ophthalmic Genet. 2018, 39, 678–683. [Google Scholar] [CrossRef]
- Johnson, A.A.; Guziewicz, K.E.; Lee, C.J.; Kalathur, R.C.; Pulido, J.S.; Marmorstein, L.Y.; Marmorstein, A.D. Bestrophin 1 and retinal disease. Prog. Retin. Eye Res. 2017, 58, 45–69. [Google Scholar] [CrossRef] [Green Version]
- Paquet-Durand, F.; Beck, S.; Michalakis, S.; Goldmann, T.; Huber, G.; Muhlfriedel, R.; Trifunovic, D.; Fischer, M.D.; Fahl, E.; Duetsch, G.; et al. A key role for cyclic nucleotide gated (CNG) channels in cGMP-related retinitis pigmentosa. Hum. Mol. Genet. 2011, 20, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Usher Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 167–170. [Google Scholar] [CrossRef]
- Tsang, S.H.; Aycinena, A.R.P.; Sharma, T. Ciliopathy: Bardet-Biedl Syndrome. Adv. Exp. Med. Biol. 2018, 1085, 171–174. [Google Scholar] [CrossRef]
- Weil, R.S.; Schrag, A.E.; Warren, J.D.; Crutch, S.J.; Lees, A.J.; Morris, H.R. Visual dysfunction in Parkinson’s disease. Brain 2016, 139, 2827–2843. [Google Scholar] [CrossRef] [Green Version]
- Girotto, G.; Vuckovic, D.; Buniello, A.; Lorente-Canovas, B.; Lewis, M.; Gasparini, P.; Steel, K.P. Expression and replication studies to identify new candidate genes involved in normal hearing function. PLoS ONE 2014, 9, e85352. [Google Scholar] [CrossRef] [PubMed]
- Booij, J.C.; Florijn, R.J.; ten Brink, J.B.; Loves, W.; Meire, F.; van Schooneveld, M.J.; de Jong, P.T.; Bergen, A.A. Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and RPGRIP1 genes in patients with juvenile retinitis pigmentosa. J. Med. Genet. 2005, 42, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, I.S.; Radziwon, A.; St. Laurent, C.D.; MacDonald, I.M. Choroideremia. Curr. Opin. Ophthalmol. 2017, 28, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gomez, C.M. Spinocerebellar [corrected] Ataxia Type 6: Molecular Mechanisms and Calcium Channel Genetics. Adv. Exp. Med. Biol. 2018, 1049, 147–173. [Google Scholar] [CrossRef]
- Stahl, J.S. Eye movements of the murine P/Q calcium channel mutant rocker, and the impact of aging. J. Neurophysiol. 2004, 91, 2066–2078. [Google Scholar] [CrossRef] [Green Version]
- Stahl, J.S.; Thumser, Z.C. Flocculus Purkinje cell signals in mouse Cacna1a calcium channel mutants of escalating severity: An investigation of the role of firing irregularity in ataxia. J. Neurophysiol. 2014, 112, 2647–2663. [Google Scholar] [CrossRef]
- Tantsis, E.M.; Gill, D.; Griffiths, L.; Gupta, S.; Lawson, J.; Maksemous, N.; Ouvrier, R.; Riant, F.; Smith, R.; Troedson, C.; et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev. Med. Child. Neurol. 2016, 58, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, K.; Mata, D.; Linn, D.M.; Linn, C.L. Neuroprotection of rat retinal ganglion cells mediated through alpha7 nicotinic acetylcholine receptors. Neuroscience 2013, 237, 184–198. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Jeon, C.J. Synaptic pattern of nicotinic acetylcholine receptor alpha7 and beta2 subunits on the direction-selective retinal ganglion cells in the postnatal mouse retina. Exp. Eye Res. 2014, 122, 54–64. [Google Scholar] [CrossRef]
- Matsumoto, H.; Shibasaki, K.; Uchigashima, M.; Koizumi, A.; Kurachi, M.; Moriwaki, Y.; Misawa, H.; Kawashima, K.; Watanabe, M.; Kishi, S.; et al. Localization of acetylcholine-related molecules in the retina: Implication of the communication from photoreceptor to retinal pigment epithelium. PLoS ONE 2012, 7, e42841. [Google Scholar] [CrossRef]
- Origlia, N.; Valenzano, D.R.; Moretti, M.; Gotti, C.; Domenici, L. Visual acuity is reduced in alpha 7 nicotinic receptor knockout mice. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkin, J.M.; Li, J.; Gilmour, G.; Mitchell, S.N.; Carter, G.; Gleason, S.D.; Seidel, W.F.; Eastwood, B.J.; McCarthy, A.; Porter, W.J.; et al. Electroencephalographic, cognitive, and neurochemical effects of LY3130481 (CERC-611), a selective antagonist of TARP-gamma8-associated AMPA receptors. Neuropharmacology 2017, 126, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.Y.; Chang, K.; Suh, Y.H.; Roche, K.W. TARP gamma-8 glycosylation regulates the surface expression of AMPA receptors. Biochem. J. 2015, 465, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Cahill, A.L.; Barta, C.; Thoreson, W.B.; Nawy, S. Elevated Pressure Increases Ca(2+) Influx Through AMPA Receptors in Select Populations of Retinal Ganglion Cells. Front. Cell Neurosci. 2018, 12, 162. [Google Scholar] [CrossRef] [Green Version]
- Almarcegui, C.; Dolz, I.; Alejos, M.V.; Fernandez, F.J.; Valdizan, J.R.; Honrubia, F.M. Pattern electroretinogram in anterior ischemic optic neuropathy. Rev. Neurol. 2001, 32, 18–21. [Google Scholar]
- Ponnalagu, D.; Rao, S.G.; Farber, J.; Xin, W.; Hussain, A.T.; Shah, K.; Tanda, S.; Berryman, M.; Edwards, J.C.; Singh, H. Molecular identity of cardiac mitochondrial chloride intracellular channel proteins. Mitochondrion 2016, 27, 6–14. [Google Scholar] [CrossRef]
- Fernandez-Martinez, L.; Letteboer, S.; Mardin, C.Y.; Weisschuh, N.; Gramer, E.; Weber, B.H.; Rautenstrauss, B.; Ferreira, P.A.; Kruse, F.E.; Reis, A.; et al. Evidence for RPGRIP1 gene as risk factor for primary open angle glaucoma. Eur. J. Hum. Genet. 2011, 19, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Raghupathy, R.K.; Zhang, X.; Liu, F.; Alhasani, R.H.; Biswas, L.; Akhtar, S.; Pan, L.; Moens, C.B.; Li, W.; Liu, M.; et al. Rpgrip1 is required for rod outer segment development and ciliary protein trafficking in zebrafish. Sci. Rep. 2017, 7, 16881. [Google Scholar] [CrossRef] [Green Version]
- Morozumi, W.; Inagaki, S.; Iwata, Y.; Nakamura, S.; Hara, H.; Shimazawa, M. Piezo channel plays a part in retinal ganglion cell damage. Exp. Eye Res. 2020, 191, 107900. [Google Scholar] [CrossRef]
- Alper, S.L. Genetic Diseases of PIEZO1 and PIEZO2 Dysfunction. Curr. Top. Membr. 2017, 79, 97–134. [Google Scholar] [CrossRef]
- Lin, Y.C.; Guo, Y.R.; Miyagi, A.; Levring, J.; MacKinnon, R.; Scheuring, S. Force-induced conformational changes in PIEZO1. Nature 2019, 573, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Ridone, P.; Vassalli, M.; Martinac, B. Piezo1 mechanosensitive channels: What are they and why are they important. Biophys. Rev. 2019, 11, 795–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.J.; Sun, D.; Jakobs, T.C. Astrocytes in the optic nerve head express putative mechanosensitive channels. Mol. Vis. 2015, 21, 749–766. [Google Scholar] [PubMed]
- Lewis, A.H.; Cui, A.F.; McDonald, M.F.; Grandl, J. Transduction of Repetitive Mechanical Stimuli by Piezo1 and Piezo2 Ion Channels. Cell Rep. 2017, 19, 2572–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CLC Genomics Workbench 20.0.4. Available online: https://digitalinsights.qiagen.com (accessed on 30 November 2020).
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Kallberg, M.; Wang, H.; Wang, S.; Peng, J.; Wang, Z.; Lu, H.; Xu, J. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 2012, 7, 1511–1522. [Google Scholar] [CrossRef] [Green Version]
- Zheng, W.; Zhang, C.; Bell, E.W.; Zhang, Y. I-TASSER gateway: A protein structure and function prediction server powered by XSEDE. Future Gener. Comput. Syst. 2019, 99, 73–85. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [Green Version]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, 38, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, K.; Wu, D.M.; Puro, D.G. Physiology of rat retinal pericytes: Modulation of ion channel activity by serum-derived molecules. J. Physiol. 1999, 521, 637–650. [Google Scholar] [CrossRef]
- Wu, D.M.; Kawamura, H.; Li, Q.; Puro, D.G. Dopamine activates ATP-sensitive K+ currents in rat retinal pericytes. Vis. Neurosci. 2001, 18, 935–940. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FEATURE | RP_2 | RP_20 | RP_21 | RP_25 | RP_32 | RP_37 | RP_38 |
|---|---|---|---|---|---|---|---|
| Age | 72 | 61 | 52 | 75 | 56 | 29 | 13 |
| Sex | F | M | F | F | M | M | M |
| Age of Onset | 4 | 8 | 4 | 24 | 10 | 9 | 3 |
| Age of First Diagnosis | 16 | 16 | 16 | 16 | 16 | 9 | b |
| Photophobia | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Night Blindness | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Color Vision | Altered | Present | Reduced | Reduced | Altered | Altered | Reduced |
| Hearing Involvement | Yes | No | No | No | Yes | Yes | Yes |
| Flashes of Lights | No | No | No | Yes | Yes | No | No |
| White Flies | Yes | No | No | Yes | Yes | No | No |
| ERG | Extinct | NA | Extinct (photopic) | Near extinction (photopic); extinct (scotopic) | Extinct (both photopic and scotopic) | Extinct (scotopic) | Quite reduced (both photopic and scotopic) |
| VEP | NA | NA | Both VEP pattern and VEP flash very reduced amplitude and normal latency in both eyes | VEP pattern with huge P100 latency increase on the right, disrupted response on the left; VEP flash with bilateral latency quite increase | NA | NA | Intact visual pathways in both eyes |
| Other | Peripheral vision, bilateral nystagmus | Altered perception of depth | Electrostimulation and magnetotherapy cleared visual perception and delayed degeneration; in relax situations visual perception is improved | Nystagmus | Tunnel vision; dizziness (with loss of balance); the intake of bananas improves visual acuity, especially the white vision | Generating cousins | Generating cousins; mild cerebellar atrophy; cone-rod dystrophy clinical diagnosis |
| FEATURE | PHENOTYPE | RP_2 | RP_20 | RP_21 | RP_25 | RP_32 | RP_37 | RP_38 |
|---|---|---|---|---|---|---|---|---|
| GUCY2D | Cone-Rod Dystrophy | rs61750173 (p.R838H) | ||||||
| rs138836357 (p.R365W) | ||||||||
| ABCA4 | rs6666652 (p.S1047I) | rs6666652 (p.S1047I) | rs6666652 (p.S1047I) | |||||
| C8orf37 | rs36096184 (p.P19S) | |||||||
| RPGRIP | rs10151259 (p.A547S) | rs10151259 (p.A547S) | ||||||
| MYO7A | Usher Syndrome | rs1052030 (p.L16Ter) | rs1052030 (p.L16Ter) | rs1052030 (p.L16Ter) | rs1052030 (p.L16Ter) | rs1052030 (p.L16Ter) | ||
| rs77625410 (Y1719C) | ||||||||
| PCDH15 | rs4935502 (p.D435A) | rs4935502 (p.D435A) | rs4935502 (p.D435A) | |||||
| USH2A | rs696723 (p.G713S) | |||||||
| SEMA4A | Retinitis Pigmentosa | rs41265017 (p.R713Q) | ||||||
| CEP290 | rs183655276 (p.D1413H) | |||||||
| AIPL1 | rs150427474 (p.R261Q) | |||||||
| TRPM1 | Congenital Stationary Night Blindness | rs138886378 (p.S157F) | ||||||
| CACNA1F | rs141159097 (p.N746T) | |||||||
| BBS2 | Bardet-Biedl Syndrome | rs4784677 (p.S70N) | rs4784677 (p.S70N) | rs4784677 (p.S70N) | rs4784677 (p.S70N) | rs4784677 (p.S70N) | ||
| CHM | Choroideremia | rs55741408 (p.L80F) |
| GENE | GO Info | RP_2 | RP_20 | RP_21 | RP_25 | RP_32 | RP_37 | RP_38 |
|---|---|---|---|---|---|---|---|---|
| ANK2 | Required for Na+/K+-ATPase, Na+/Ca2+ exchanger and Beta-2-Spectrin expression; abundant in rods | I825T | ||||||
| CACNA1A | The channel activity is directed by the pore-forming alpha-1 subunit, whereas the others act as auxiliary subunits. The isoform alpha-1A gives rise to P and/or Q-type calcium currents | Q792_Q800del | ||||||
| CACNG8 | Modulates Ca2+ L-type channel activity and AMPAR opening | G327C; G361V | ||||||
| CHRNA7 | Subunit of nicotinic ACh channel-receptor; it forms a homo-oligomeric channel with high permeability to Ca2+ | L166fs | L166fs | |||||
| CLIC5 | Constitutes channel with low-selectivity, also for Cl− | S106ter | S106ter | S106ter | S106ter | |||
| CNGB1 | Subunit of nonselective cationic ion channels mediated by cyclic nucleotides; abundant in rods | E370_E371del | ||||||
| CNGB3 | Beta subunit of a cationic ion channel mediated by cyclic nucleotides; abundant in cones | R781C | R781C | R781C | R781C | R781C | ||
| DRD4 | D4-subtype of dopamine receptors; abundant in retinal ganglion cells | D288G | ||||||
| LRRK2 | Serine/threonine kinase which phosphorylates proteins involved into neuronal plasticity, autophagy and vesicular trafficking | P1262A | ||||||
| PIEZO1 | Ion channel which boosts the intracellular entry of Ca2+ | V250A; K1878del |
| Gene | Wt/Mutated | MW [g/moL] | Net Charge [pH = 7] | pI | Average Hydropathy | Aliphatic Index | A₂₈₀ (ox.) | A₂₈₀ (red.) | ε₂₈₀ [M−¹ cm−¹] | Instability Index | Features |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ANK2 | Wt | 165,738.9 | −188.1 | 4.32 | −0.88 | 60.79 | 0.38 | 0.37 | 61,770 | 72.04 | EFO, CA, MG, OHX, MN, MG, NDP, MYR, CA, DCA, 43 helices, 62 strands |
| I825T | 165,726.94 | −188.01 | 4.32 | −0.88 | 60.53 | 0.38 | 0.37 | 61,770 | 71.82 | 86 helices, 124 strands, I825T | |
| CACNA1A | Wt | 225,243.55 | 34.65 | 9.14 | −0.59 | 68.01 | 0.99 | 0.99 | 222,890 | 53.1 | 9SL, ANP, III, MGE, Y01, CA, MG, 6OU, 61 helices, 4 strands |
| Q792_Q800del | 218,047.31 | 36.67 | 9.22 | −0.60 | 68.10 | 0.97 | 0.97 | 211,430 | 52.29 | 9SL, III, CA, Y01, ANP, UUU, MG, MGE, 9Z9, 6OU, 74 helices | |
| CACNG8 | Wt | 43,312.87 | 11.22 | 9.34 | −0.10 | 77.93 | 1.09 | 1.08 | 46,870 | 57.88 | ACD, BCL, XE, III, ZN, CVM, MG, NUC, CA, CA, 11 helices, 5 strands |
| G327C; G361V | 43,401.04 | 11.16 | 9.29 | −0.08 | 78.61 | 1.09 | 1.08 | 46,870 | 57.21 | 22 helices, 10 strands, G327C, G361V | |
| CHRNA7 | Wt | 56,449.44 | −6.91 | 6.02 | 0.08 | 95.10 | 1.76 | 1.74 | 98,320 | 44.56 | III, MLK, DSF, ZY5, EPJ, IVM, TC9, 10 helices, 12 strands |
| L166fs | 23,812.21 | 0.21 | 6.95 | −0.31 | 86.44 | 2.71 | 2.7 | 64,400 | 47.42 | III, CU9, MLK, PLC, V11, 9Z0, 3 helices, 10 strands | |
| CLIC5 | Wt | 46,502.65 | −29.41 | 4.71 | −0.69 | 73.32 | 0.95 | 0.94 | 43,780 | 45.83 | GSH, MNB, ASC, P10, GTX, GDS, CA, 12 helices, 6 strands |
| S106ter | 12,409.07 | −22.62 | 3.97 | −1.21 | 60.66 | 1.20 | 1.20 | 14,900 | 56.84 | FE2, CA, MN, FAD, OXD, OXY, ZN, ACT, MN3, 1 helix, 3 strands | |
| CNGB1 | Wt | 139,677.76 | −78.93 | 4.75 | −0.60 | 75.08 | 1.15 | 1.14 | 159,170 | 65.92 | CMP, ANP, 6ZL, CLA, PGW, III, CA, SF4, MG, 30 helices, 12 strands |
| E370_E371del | 139,419.53 | −76.93 | 4.77 | −0.59 | 75.20 | 1.15 | 1.14 | 159,170 | 65.49 | CMP, B73, ANP, PGW, IAC, K, CLA, MG, CA, CH1, 28 helices, 11 strands | |
| CNGB3 | Wt | 92,166.52 | 3.64 | 8.06 | −0.51 | 82.81 | 1.09 | 1.09 | 100,160 | 43.43 | CMP, PGW, CA, SF4, 78M, ANP, K, III, CLA, SF4, 29 helices, 13 strands |
| R781C | 92,113.47 | 2.58 | 7.81 | −0.50 | 82.81 | 1.09 | 1.09 | 100,160 | 43.26 | 58 helices, 26 strands, R781C | |
| DRD4 | Wt | 43,901.46 | 14.89 | 9.24 | 0.30 | 96.01 | 1.02 | 0.99 | 43,430 | 49.26 | AQD, ERC, NA, CLR, 1WV, CLR, 2CV, 2CV, SOG, CLR, 17 helices |
| D288G | 43,843.43 | 15.88 | 9.31 | 0.31 | 96.01 | 1.02 | 0.99 | 43,430 | 48.81 | 34 helices, D288G | |
| LRRK2 | Wt | 224,673.52 | −31.59 | 6.06 | −0.11 | 104.81 | 0.73 | 0.72 | 161,120 | 45.81 | CA, CLA, III, III, SE, UUU, MG, UUU, UUU, III, 98 helices, 4 strands |
| P1262A | 224,647.49 | −30.59 | 6.06 | −0.11 | 104.85 | 0.73 | 0.72 | 161,120 | 45.65 | 196 helices, 8 strands, P1262A | |
| PIEZO1 | Wt | 224,984.30 | −1.22 | 7.07 | 0.06 | 101.72 | 1.66 | 1.65 | 370,820 | 51.95 | ANP, 9SL, MGE, NUC, Y01, MG, III, 9Z9, CA, GRG, 79 helices, 4 strands |
| V250A; K1878del | 226,789.15 | −4.13 | 6.85 | 0.06 | 101.34 | 1.65 | 1.64 | 370,820 | 51.55 | MGE, III, BCL, ANP, CA, P10, GRG, CLA, MG, 104 helices, 2 strands |
| GENE | VARIANT | LIGAND BINDING SITE (Wt) [AA] | LIGAND BINDING SITE (Mut) [AA] | NOTES |
|---|---|---|---|---|
| ANK2 | I825T | EF0: 1498 Mg: 1499 F2A: 1500 NDP: 1501 Ca: 1502 | EF0: 1498 Mg: 1499 F2A: 1500 NDP: 1501 Ca: 1502 | Binding sites for all predicted ligands identical for wild and mutated proteins |
| CACNA1A | Q792_Q800del | 9SL: 1997 ANP: 1998 Y01: 1999 Ca: 2000 Mg: 2001 6OU: 2002 | 9SL: 1937 Ca: 1938 Y01: 1939 ANP: 1940 9Z9: 1941 6OU: 1942 | Ca2+ binding sites almost shifted, deletion of a Mg2+ binding site in mutated protein |
| CACNG8 | G327C; G361V | ACD: 426 BCL: 427 Ca: 430 XE: 428 Zn: 429 | ACD: 426 BCL: 427 Ca: 430 XE: 428 Zn: 429 | Binding sites for all predicted ligands identical for wild and mutated proteins |
| CHRNA7 | L166fs | NH2: 503, 506, 509 MLK: 504 DSF: 505 ZY5: 507 EPJ: 508 IVM: 510 TC9: 511 | NH2: 206 Cu9: 207 9Z0: 208–209 | Deletion of several ligand binding sites in mutated protein |
| CLIC5 | S106ter | GSH: 411, 415 GTT: 412 MNB: 413 ASC: 414 GTX: 416 GDS: 417 Ca: 418 | Fe2: 107 Mn: 108 Ca: 109 FAD: 110 OXD: 111 OXY: 112 Zn: 113 Mn3: 114 | Ca2+ binding sites almost shifted, creation of a several new ion binding sites in mutated protein |
| CNGB1 | E370_E371del | Unknown: 1252-1264 ANP: 1265-1266 6ZL: 1267 CLA: 1268 PGW: 1269 SF4: 1270 Mg: 1271 | B73: 1250 ANP: 1251 PGW: 1252 IAC: 1253 K: 1254 CLA: 1255 Mg: 1256 Ca: 1257 CH1: 1258 | Mg2+ binding site almost shifted, creation of a K+ and Ca2+ binding sites in mutated protein |
| CNGB3 | R781C | PGW: 810 Ca: 811 SF4: 812, 817 78M: 813 ANP: 814 K: 815 CLA: 816 | PGW: 810 Ca: 811 SF4: 812, 817 78M: 813 ANP: 814 K: 815 CLA: 816 | Binding sites for all predicted ligands identical for wild and mutated proteins |
| DRD4 | D288G | AQD: 420 ERC: 421 Na: 422 CLR: 423, 425, 427 1WV: 424 WHJ: 426 | AQD: 420 ERC: 421 Na: 422 CLR: 423, 425, 427 1WV: 424 WHJ: 426 | Binding sites for all predicted ligands identical for wild and mutated proteins |
| LRRK2 | P1262A | Unknown: 1981-1984 | Unknown: / | Variant near Mg2+ binding site, far from Se and Ca2+ ones |
| PIEZO1 | V250A; K1878del | ANP: 1981 9SL: 1982 Y01: 1983 9Z9: 1984 Ca: 1985 | Unknown: 1999-2000 BCL: 2001 ANP: 2002 Ca: 2003 CLA: 2004 Mg: 2005 | Ca2+ binding sites almost shifted, creation of a Mg2+ binding site in mutated protein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donato, L.; Scimone, C.; Alibrandi, S.; Abdalla, E.M.; Nabil, K.M.; D’Angelo, R.; Sidoti, A. New Omics—Derived Perspectives on Retinal Dystrophies: Could Ion Channels-Encoding or Related Genes Act as Modifier of Pathological Phenotype? Int. J. Mol. Sci. 2021, 22, 70. https://doi.org/10.3390/ijms22010070
Donato L, Scimone C, Alibrandi S, Abdalla EM, Nabil KM, D’Angelo R, Sidoti A. New Omics—Derived Perspectives on Retinal Dystrophies: Could Ion Channels-Encoding or Related Genes Act as Modifier of Pathological Phenotype? International Journal of Molecular Sciences. 2021; 22(1):70. https://doi.org/10.3390/ijms22010070
Chicago/Turabian StyleDonato, Luigi, Concetta Scimone, Simona Alibrandi, Ebtesam Mohamed Abdalla, Karim Mahmoud Nabil, Rosalia D’Angelo, and Antonina Sidoti. 2021. "New Omics—Derived Perspectives on Retinal Dystrophies: Could Ion Channels-Encoding or Related Genes Act as Modifier of Pathological Phenotype?" International Journal of Molecular Sciences 22, no. 1: 70. https://doi.org/10.3390/ijms22010070