
Longitudinal Changes in Diet Cause Repeatable and Largely Reversible Shifts in Gut Microbial Communities of Laboratory Mice and Are Observed across Segments of the Entire Intestinal Tract

 , ,
, , 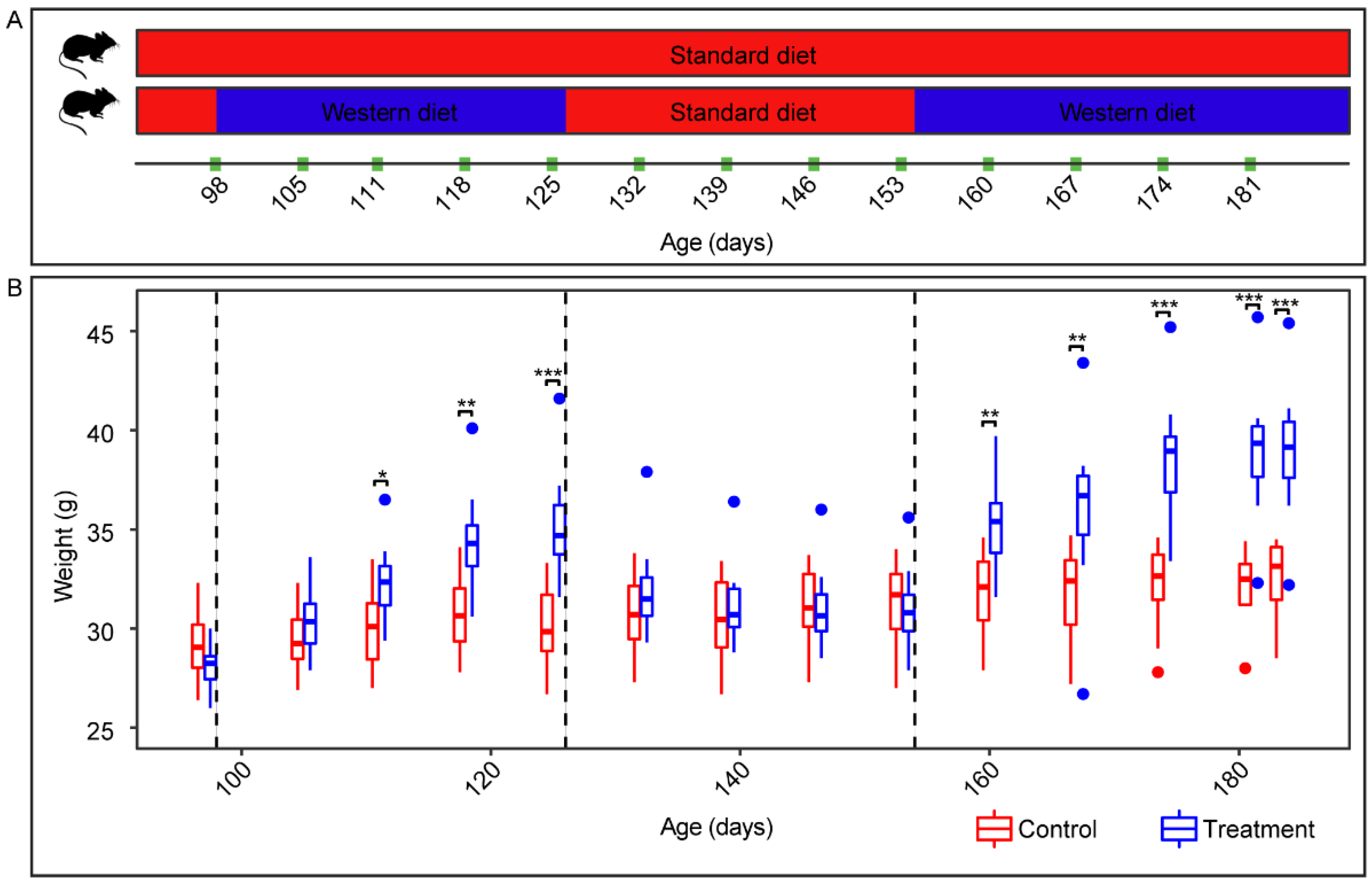{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effects of a Western Diet on Animal Weight and Gastrointestinal Tract Length
2.2. Effects of Diet-Switches on the Fecal Microbiota
2.3. Diet-Dependent Microbiota Differences Are Observable along the Alimentary Tract
2.4. Taxonomic Compositional Changes along the GIT and in Fecal Samples of WD-Fed Mice
2.5. Predictive Metagenomic Profiling in Segments of Mice Fed Standard and Western Diets
2.6. Discussion
3. Materials and Methods
3.1. Diet Experiment, Fecal Sampling, and Gastrointestinal Segments Harvest
3.2. DNA Extraction, Indexing, and Amplicon Sequencing
3.3. Amplicon Sequences Processing, Microbiota and Predicted Metagenome Analyses
3.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hugenholtz, F.; de Vos, W.M. Mouse models for human intestinal microbiota research: A critical evaluation. Cell Mol. Life Sci. 2018, 75, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, S.W.; Calabrese, G.; Kolling, G.L.; Platts-Mills, J.; Freire, R.; Alcantara-Warren, C.; Liu, B.; Sartor, R.B.; Guerrant, R.L. Murine model of Clostridium difficile infection with aged gnotobiotic C57BL/6 mice and a BI/NAP1 strain. J. Infect. Dis. 2010, 202, 1708–1712. [Google Scholar] [CrossRef]
- Bloom, S.M.; Bijanki, V.N.; Nava, G.M.; Sun, L.; Malvin, N.P.; Donermeyer, D.L.; Dunne, W.M., Jr.; Allen, P.M.; Stappenbeck, T.S. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 2011, 9, 390–403. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14–16ra14. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Chen, D.; Zhang, J.N.; Lv, X.; Wang, K.; Duan, L.P.; Nie, Y.; Wu, X.L. Bacterial community mapping of the mouse gastrointestinal tract. PLoS ONE 2013, 8, e74957. [Google Scholar] [CrossRef]
- Martinez-Guryn, K.; Leone, V.; Chang, E.B. Regional diversity of the gastrointestinal microbiome. Cell Host Microbe 2019, 26, 314–324. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Lesker, T.R.; Hitch, T.C.A.; Galvez, E.J.C.; Smit, N.; Neuhaus, K.; Wang, J.; Baines, J.F.; Abt, B.; Stecher, B.; et al. Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 2019, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Yang, B.; Ye, C.; Yan, B.; He, X.; Xing, K. Assessing the influence of dietary history on gut microbiota. Curr. Microbiol. 2019, 76, 237–247. [Google Scholar] [CrossRef]
- Myles, I.A.; Fontecilla, N.M.; Janelsins, B.M.; Vithayathil, P.J.; Segre, J.A.; Datta, S.K. Parental dietary fat intake alters offspring microbiome and immunity. J. Immunol. 2013, 191, 3200–3209. [Google Scholar] [CrossRef]
- Carmody, R.N.; Gerber, G.K.; Luevano, J.M., Jr.; Gatti, D.M.; Somes, L.; Svenson, K.L.; Turnbaugh, P.J. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe 2015, 17, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wang, L.; Yang, D.; Li, L.; Togo, J.; Wu, Y.; Liu, Q.; Li, B.; Li, M.; Wang, G.; et al. Dietary fat, but not protein or carbohydrate, regulates energy intake and causes adiposity in mice. Cell Metab. 2018, 28, 415–431. [Google Scholar] [CrossRef] [PubMed]
- Naya, D.E.; Karasov, W.H.; Bozinovic, F. Phenotypic plasticity in laboratory mice and rats: A meta-analysis of current ideas on gut size flexibility. Evol. Ecol. Res. 2007, 9, 1363–1374. [Google Scholar]
- Dalby, M.J.; Ross, A.W.; Walker, A.W.; Morgan, P.J. Dietary uncoupling of gut microbiota and energy harvesting from obesity and glucose tolerance in mice. Cell Rep. 2017, 21, 1521–1533. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Backhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, M.; Pang, X.; Zhao, Y.; Wang, L.; Zhao, L. Structural resilience of the gut microbiota in adult mice under high-fat dietary perturbations. ISME J. 2012, 6, 1848–1857. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Backhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Chassaing, B.; Miles-Brown, J.; Pellizzon, M.; Ulman, E.; Ricci, M.; Zhang, L.; Patterson, A.D.; Vijay-Kumar, M.; Gewirtz, A.T. Lack of soluble fiber drives diet-induced adiposity in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G528–G541. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef]
- Hoy, Y.E.; Bik, E.M.; Lawley, T.D.; Holmes, S.P.; Monack, D.M.; Theriot, J.A.; Relman, D.A. Variation in taxonomic composition of the fecal microbiota in an inbred mouse strain across individuals and time. PLoS ONE 2015, 10, e0142825. [Google Scholar] [CrossRef]
- Low, A.; Soh, M.; Miyake, S.; Seedorf, H. Host-age prediction from fecal microbiome composition in laboratory mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Suzuki, T.A.; Nachman, M.W. Spatial heterogeneity of gut microbial composition along the gastrointestinal tract in natural populations of house mice. PLoS ONE 2016, 11, e0163720. [Google Scholar] [CrossRef]
- Ericsson, A.C.; Gagliardi, J.; Bouhan, D.; Spollen, W.G.; Givan, S.A.; Franklin, C.L. The influence of caging, bedding, and diet on the composition of the microbiota in different regions of the mouse gut. Sci. Rep. 2018, 8, 4065. [Google Scholar] [CrossRef]
- Ebino, K.Y.; Suwa, T.; Kuwabara, Y.; Saito, T.R.; Takahashi, K.W. Lifelong coprophagy in male mice. Exp. Anim. 1987, 36, 273–276. [Google Scholar] [CrossRef]
- Bogatyrev, S.R.; Rolando, J.C.; Ismagilov, R.F. Self-reinoculation with fecal flora changes microbiota density and composition leading to an altered bile-acid profile in the mouse small intestine. Microbiome 2020, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Bisanz, J.E.; Upadhyay, V.; Turnbaugh, J.A.; Ly, K.; Turnbaugh, P.J. Meta-analysis reveals reproducible gut microbiome alterations in response to a high-fat diet. Cell Host Microbe 2019, 26, 265–272. [Google Scholar] [CrossRef]
- Park, J.K.; Chang, D.H.; Rhee, M.S.; Jeong, H.; Song, J.; Ku, B.J.; Kim, S.B.; Lee, M.; Kim, B.C. Heminiphilus faecis gen. nov., sp. nov., a member of the family Muribaculaceae, isolated from mouse faeces and emended description of the genus Muribaculum. Antonie Van Leeuwenhoek 2021, 114, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Lagkouvardos, I.; Pukall, R.; Abt, B.; Foesel, B.U.; Meier-Kolthoff, J.P.; Kumar, N.; Bresciani, A.; Martínez, I.; Just, S.; Ziegler, C. The Mouse Intestinal Bacterial Collection (miBC) provides host-specific insight into cultured diversity and functional potential of the gut microbiota. Nat. Microbiol. 2016, 1, 16131. [Google Scholar] [CrossRef] [PubMed]
- Miyake, S.; Ding, Y.; Soh, M.; Low, A.; Seedorf, H. Cultivation and description of Duncaniella dubosii sp. nov., Duncaniella freteri sp. nov. and emended description of the species Duncaniella muris. Int. J. Syst. Evol. Microbiol. 2020, 70, 3105–3110. [Google Scholar] [CrossRef]
- Miyake, S.; Ding, Y.; Soh, M.; Seedorf, H. Complete genome sequence of Duncaniella muris strain B8, isolated from the feces of C57/BL6 mice. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Miyake, S.; Ding, Y.; Soh, M.; Low, A.; Seedorf, H. Muribaculum gordoncarteri sp. nov., an anaerobic bacterium from the faeces of C57BL/6J mice. Int. J. Syst. Evol. Microbiol. 2020, 70, 4725–4729. [Google Scholar] [CrossRef] [PubMed]
- Seedorf, H.; Griffin, N.W.; Ridaura, V.K.; Reyes, A.; Cheng, J.; Rey, F.E.; Smith, M.I.; Simon, G.M.; Scheffrahn, R.H.; Woebken, D.; et al. Bacteria from diverse habitats colonize and compete in the mouse gut. Cell 2014, 159, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Smolin, J.; Nayak, J.; Ayala, J.E.; Scott, D.A.; Peterson, S.N.; Freeze, H.H. Mannose alters gut microbiome, prevents diet-induced obesity, and improves host metabolism. Cell Rep. 2018, 24, 3087–3098. [Google Scholar] [CrossRef]
- Chang, D.-H.; Rhee, M.-S.; Ahn, S.; Bang, B.-H.; Oh, J.E.; Lee, H.K.; Kim, B.-C. Faecalibaculum rodentium gen. nov., sp. nov., isolated from the faeces of a laboratory mouse. Antonie Van Leeuwenhoek 2015, 108, 1309–1318. [Google Scholar] [CrossRef]
- Ericsson, A.C.; Davis, J.W.; Spollen, W.; Bivens, N.; Givan, S.; Hagan, C.E.; McIntosh, M.; Franklin, C.L. Effects of vendor and genetic background on the composition of the fecal microbiota of inbred mice. PLoS ONE 2015, 10, e0116704. [Google Scholar] [CrossRef]
- Drozdowski, L.A.; Thomson, A.B. Intestinal sugar transport. World J. Gastroenterol. 2006, 12, 1657–1670. [Google Scholar] [CrossRef]
- Lesker, T.R.; Durairaj, A.C.; Galvez, E.J.C.; Lagkouvardos, I.; Baines, J.F.; Clavel, T.; Sczyrba, A.; McHardy, A.C.; Strowig, T. An integrated metagenome catalog reveals new insights into the murine gut microbiome. Cell Rep. 2020, 30, 2909–2922. [Google Scholar] [CrossRef]
- Jiang, P.; Green, S.J.; Chlipala, G.E.; Turek, F.W.; Vitaterna, M.H. Reproducible changes in the gut microbiome suggest a shift in microbial and host metabolism during spaceflight. Microbiome 2019, 7, 113. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, N.; Du, M.-X.; Sun, Y.-T.; Wang, K.; Wang, Y.-J.; Li, D.-H.; Yu, H.-Y.; Song, Y.; Bai, B.-B. The Mouse Gut Microbial Biobank expands the coverage of cultured bacteria. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Million, M.; Tomas, J.; Wagner, C.; Lelouard, H.; Raoult, D.; Gorvel, J.-P. New insights in gut microbiota and mucosal immunity of the small intestine. Human Microbiome J. 2018, 7, 23–32. [Google Scholar] [CrossRef]
- Ruehl-Fehlert, C.; Kittel, B.; Morawietz, G.; Deslex, P.; Keenan, C.; Mahrt, C.R.; Nolte, T.; Robinson, M.; Stuart, B.P.; Deschl, U.; et al. Revised guides for organ sampling and trimming in rats and mice—Part 1. Exp. Toxicol. Pathol. 2003, 55, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Rius, A.G.; Kittelmann, S.; Macdonald, K.A.; Waghorn, G.C.; Janssen, P.H.; Sikkema, E. Nitrogen metabolism and rumen microbial enumeration in lactating cows with divergent residual feed intake fed high-digestibility pasture. J. Dairy Sci. 2012, 95, 5024–5034. [Google Scholar] [CrossRef] [PubMed]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 1091. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2007, 36, D623–D631. [Google Scholar] [CrossRef]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.M.; Harris, H.M.; Mattarelli, P.; O’Toole, P.W.; Pot, B.; Vandamme, P.; Walter, J. A taxonomic note on the genus Lactobacillus: Description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 2782–2858. [Google Scholar] [CrossRef]
- Vazquez-Baeza, Y.; Gonzalez, A.; Smarr, L.; McDonald, D.; Morton, J.T.; Navas-Molina, J.A.; Knight, R. Bringing the dynamic microbiome to life with animations. Cell Host Microbe 2017, 21, 7–10. [Google Scholar] [CrossRef]
- Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin, Germany, 2016. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed]
- Bisanz, J.E. qiime2R: Importing QIIME2 Artifacts and Associated Data into R Sessions, v0.99. Available online: https://github.com/jbisanz/qiime2R (accessed on 10 May 2021).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J. Welcome to the Tidyverse. J. Open Sour. Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual comparisons by ranking methods. Biometrics 1945, 1, 80–83. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbo, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Low, A.; Soh, M.; Miyake, S.; Aw, V.Z.J.; Feng, J.; Wong, A.; Seedorf, H. Longitudinal Changes in Diet Cause Repeatable and Largely Reversible Shifts in Gut Microbial Communities of Laboratory Mice and Are Observed across Segments of the Entire Intestinal Tract. Int. J. Mol. Sci. 2021, 22, 5981. https://doi.org/10.3390/ijms22115981
Low A, Soh M, Miyake S, Aw VZJ, Feng J, Wong A, Seedorf H. Longitudinal Changes in Diet Cause Repeatable and Largely Reversible Shifts in Gut Microbial Communities of Laboratory Mice and Are Observed across Segments of the Entire Intestinal Tract. International Journal of Molecular Sciences. 2021; 22(11):5981. https://doi.org/10.3390/ijms22115981
Chicago/Turabian StyleLow, Adrian, Melissa Soh, Sou Miyake, Vanessa Zhi Jie Aw, Jian Feng, Asher Wong, and Henning Seedorf. 2021. "Longitudinal Changes in Diet Cause Repeatable and Largely Reversible Shifts in Gut Microbial Communities of Laboratory Mice and Are Observed across Segments of the Entire Intestinal Tract" International Journal of Molecular Sciences 22, no. 11: 5981. https://doi.org/10.3390/ijms22115981