
Product Distributions of Cytochrome P450 OleTJE with Phenyl-Substituted Fatty Acids: A Computational Study


1
Manchester Institute of Biotechnology, The University of Manchester, Manchester M1 7DN, UK
2
Department of Chemical Engineering and Analytical Science, The University of Manchester, Manchester M13 9PL, UK
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(13), 7172; https://doi.org/10.3390/ijms22137172
Submission received: 14 June 2021
/
Revised: 29 June 2021
/
Accepted: 29 June 2021
/
Published: 2 July 2021
(This article belongs to the Special Issue Biocatalysis, a Life Companion for Green Chemistry: Biomolecular Aspects of Bioprocesses)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:There are two types of cytochrome P450 enzymes in nature, namely, the monooxygenases and the peroxygenases. Both enzyme classes participate in substrate biodegradation or biosynthesis reactions in nature, but the P450 monooxygenases use dioxygen, while the peroxygenases take H2O2 in their catalytic cycle instead. By contrast to the P450 monooxygenases, the P450 peroxygenases do not require an external redox partner to deliver electrons during the catalytic cycle, and also no external proton source is needed. Therefore, they are fully self-sufficient, which affords them opportunities in biotechnological applications. One specific P450 peroxygenase, namely, P450 OleTJE, reacts with long-chain linear fatty acids through oxidative decarboxylation to form hydrocarbons and, as such, has been implicated as a suitable source for the biosynthesis of biofuels. Unfortunately, the reactions were shown to produce a considerable amount of side products originating from Cα and Cβ hydroxylation and desaturation. These product distributions were found to be strongly dependent on whether the substrate had substituents on the Cα and/or Cβ atoms. To understand the bifurcation pathways of substrate activation by P450 OleTJE leading to decarboxylation, Cα hydroxylation, Cβ hydroxylation and Cα–Cβ desaturation, we performed a computational study using 3-phenylpropionate and 2-phenylbutyrate as substrates. We set up large cluster models containing the heme, the substrate and the key features of the substrate binding pocket and calculated (using density functional theory) the pathways leading to the four possible products. This work predicts that the two substrates will react with different reaction rates due to accessibility differences of the substrates to the active oxidant, and, as a consequence, these two substrates will also generate different products. This work explains how the substrate binding pocket of P450 OleTJE guides a reaction to a chemoselectivity.
1. Introduction
With the advent of knowledge on biochemical reaction processes and mechanisms, the use of biosystems in engineering processes has increased rapidly. In particular, enzymes are now commonly applied in industrial processes related to food production as well as the synthesis of fine-chemicals. One area of great potential for enzymes is the field of biofuels, where they can be utilized for the synthesis of hydrocarbons. Interestingly, in nature, there are several enzymes involved in the conversion of alcohols or fatty acids into hydrocarbons [1,2,3,4,5,6], and, as such, there is potential to use these for biotechnical applications. Most of these enzymes are metalloenzymes, where a reaction on either an iron or copper center takes place. Metalloenzymes are highly versatile in nature and catalyze substrate oxidation and reduction reactions efficiently [7,8,9]. In particular, the oxygen-activating mononuclear iron-containing enzymes are useful in this respect. They have been assigned to two different categories, namely, the heme monooxygenases [10,11,12,13,14,15,16,17,18] and the nonheme iron dioxygenases [19,20,21,22,23,24,25,26], where the former group includes the cytochromes P450 involved in biosynthesis and biodegradation processes.
The cytochromes P450 are heme enzymes that either take O2 and react as monooxygenases or use H2O2 and operate as peroxygenases. P450 monooxygenases react dioxygen on an iron (III)-heme center to form an iron (IV)-oxo heme cation radical species called Compound I (CpdI) with the help of two external electrons (from redox partners) and two protons from the solvent according to the mechanism shown in Scheme 1. CpdI is a versatile catalyst that reacts with aliphatic chains through substrate hydroxylation or desaturation, or with olefins and arenes to form epoxides and phenols. In addition to the P450 monooxygenases, there are several P450s that use H2O2 instead of dioxygen and therefore act as peroxygenases. These also form CpdI in their catalytic cycle but do not need external electrons and protons as they are provided by H2O2 (bottom reaction in Scheme 1). As such, these P450 peroxygenases are self-sufficient and more suitable for biotechnological applications than the P450 monooxygenases. The P450 peroxygenases were reported to take long-chain fatty acids as substrates and react them through an oxidative decarboxylation to form terminal olefins [27,28,29,30,31,32,33,34,35,36,37].
P450 peroxygenases were also shown to react with a variety of small carboxylic acids; however, the product distributions were markedly different between the various substrates [36]. Thus, Pickl et al. [36] showed that P450 OleTJE reacts with linear fatty acids, and, e.g., a substrate with the chain length of 13 carbon atoms produced dominant decarboxylation products to form the C12 olefin with a minor amount of Cβ hydroxylation. By contrast, using R- or S-2-methylbutyrate instead, P450 OleTJE converted the substrate to the Cα-hydroxylated product or caused desaturation of the Cα–Cβ bond. Computational modeling on the mechanism of the latter reaction showed that the substrate was able to bind in various conformations that led to different reaction channels and products that are close in energy [36,38]. Yet, when P450 OleTJE was fed with 2-methyl-3-phenylpropionate, the reaction products corresponded to decarboxylation and Cα hydroxylation instead [36], as shown in Scheme 2. By contrast, the reaction of P450 OleTJE with 2-phenylbutyrate gave a major reaction channel for Cα hydroxylation and a minor amount of Cα–Cβ desaturation. Clearly, the reaction products of substrate activation by P450 OleTJE are highly dependent on the size and shape of the substrate, and the origins of these product distributions remain unclear. Therefore, we decided to conduct another computational study into substrate activation by P450 OleTJE, investigate substrates with phenyl substituents and find out how P450 OleTJE would activate these. In particular, no computational studies have been reported on fatty acids with phenyl side chains that create bulkier substrates than linear long-chain fatty acids and hence may not fit the substrate binding pocket well.
2. Results
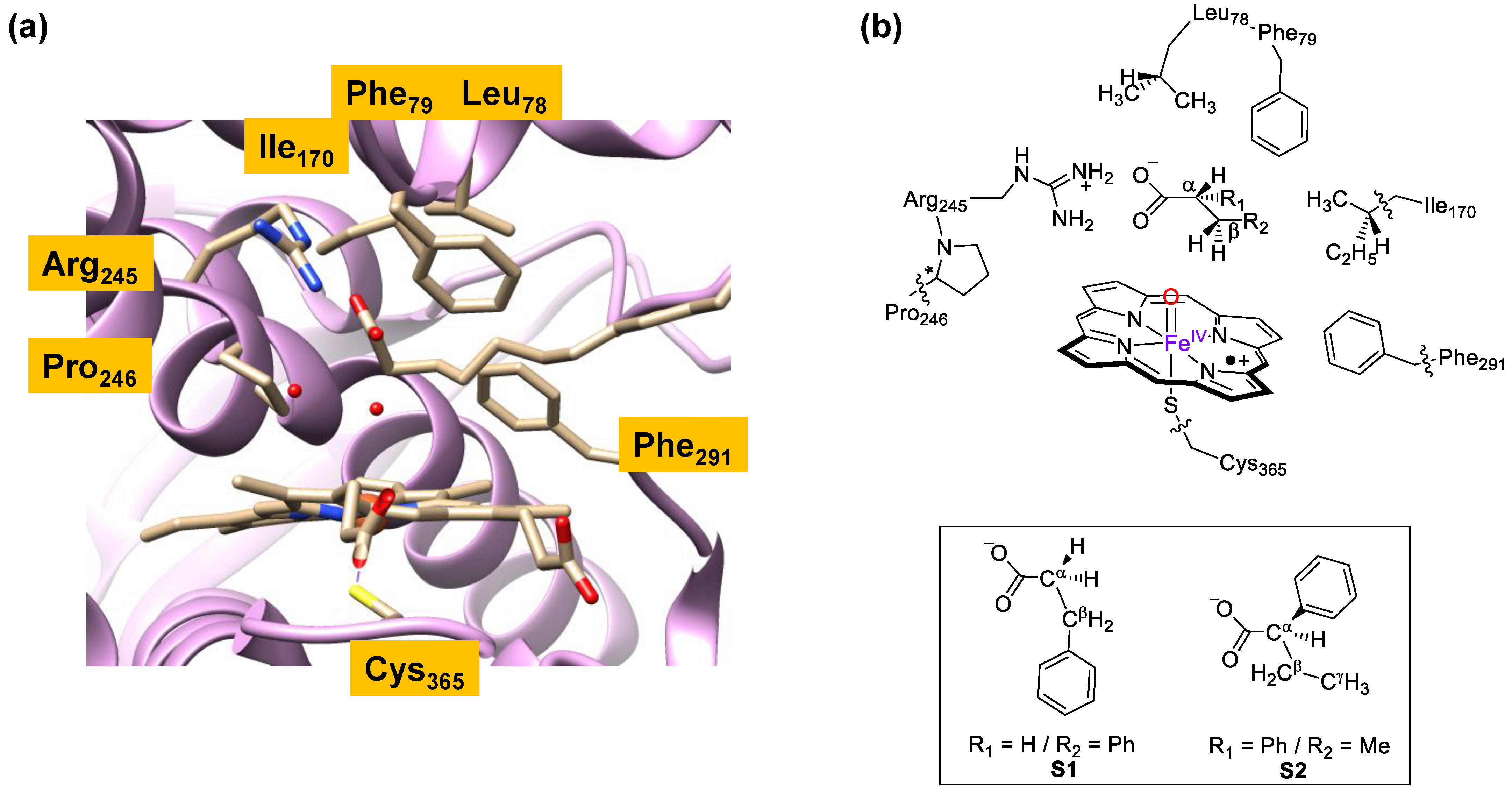
To understand the mechanism of substrate activation by P450 OleTJE enzymes and, in particular, how the enzymes react with fatty acid substrates with phenyl substituents, we decided to create a series of cluster models and study the possible reaction pathways for fatty acid activation by CpdI models. Several previous studies covered the H2O2 activation on a heme center to form CpdI [39,40,41,42], which is the active oxidant that performs the catalysis. Hence, we will focus on the mechanism starting from CpdI and a substrate only. Previous studies of ours showed that large QM cluster models of well over 150 atoms represent the oxidant and substrate binding pockets of enzymes highly accurately and give results at par with QM/MM methods [43,44,45,46]. Therefore, a large QM cluster model of the substrate-bound P450 OleT models was set up. The substrate-bound structure of P450 OleTJE was characterized crystallographically by Belcher et al. [47], and an extract of the 4L40 protein databank file [48] (pdb) is shown in Figure 1. As it can be seen, the heme is linked to the protein through a thiolate linkage of Cys365. The substrate binding pocket is mostly lined with aromatic and aliphatic amino acids including the side chains of Leu78, Phe79, Ile170, Pro246 and Phe291. In the substrate binding pocket, the carboxylate of the icosanoic acid substrate forms a salt bridge with the side chain of Arg245.
To understand the substrate activation mechanism, we set up cluster models using previously reported methods that take key hydrogen bonding and polar residues into consideration [49,50]. In order to do so, we removed the icosanoic acid from the pdb and docked an alternative substrate manually into the pocket. These substrates lack the long aliphatic tail of icosanoic acid, and hence the models we created only included protein residues within the vicinity of the new substrates. In particular, we created two reactant models, namely, ReS1 and ReS2, which have the same protein structure but contain a different substrate, i.e., either S1 or S2, Figure 1b. Thus, substrate S1 is 3-phenylpropionate and substrate S2 is 2-phenylbutyrate. Model ReS1 has 162 atoms in total, whereas model ReS2 contains 165 atoms. Both systems have a neutral charge and were calculated in the lowest energy doublet and quartet spin states, as identified with the superscript of 2 or 4 next to the labels. Following previous experience with substrate binding into the active site of P450 OleTJE [36], we made sure the substrate forms a double hydrogen bonding interaction and salt bridge of its carboxylate group with the methylguanidinium group of Arg245. In addition, the model contains the heme with all ring substituents truncated to hydrogen atoms and the axial Cys365 residue as thiolate. The substrate binding pocket contains the peptide dimers Leu78-Phe79 and Arg245-Pro246 as well as the side chains of residues Ile170 and Phe291. A previous study used the same peptide and active site model but with hexanoate as substrate [38], whereby the model was validated against high-level quantum mechanics/molecular mechanics (QM/MM) calculations [51,52] and large cluster models [53]. These studies found very good agreement of the calculations of energy landscapes for a reaction mechanism, as obtained with either cluster models or QM/MM. In addition, a range of computational methods were tested, namely, several density functional theory methods and basis sets, and all approaches came to the same conclusions and predicted similar rate constants. As such, we will use large cluster models here with the B3LYP method in combination with a double-ζ quality basis set on all atoms, i.e., LACVP basis set with core potential on iron and 6-31G on the rest of the atoms, while a solvent model with a dielectric constant of ε = 5.7 was included in the geometry optimizations, as well, as that enables a comparison with previous work in the field.
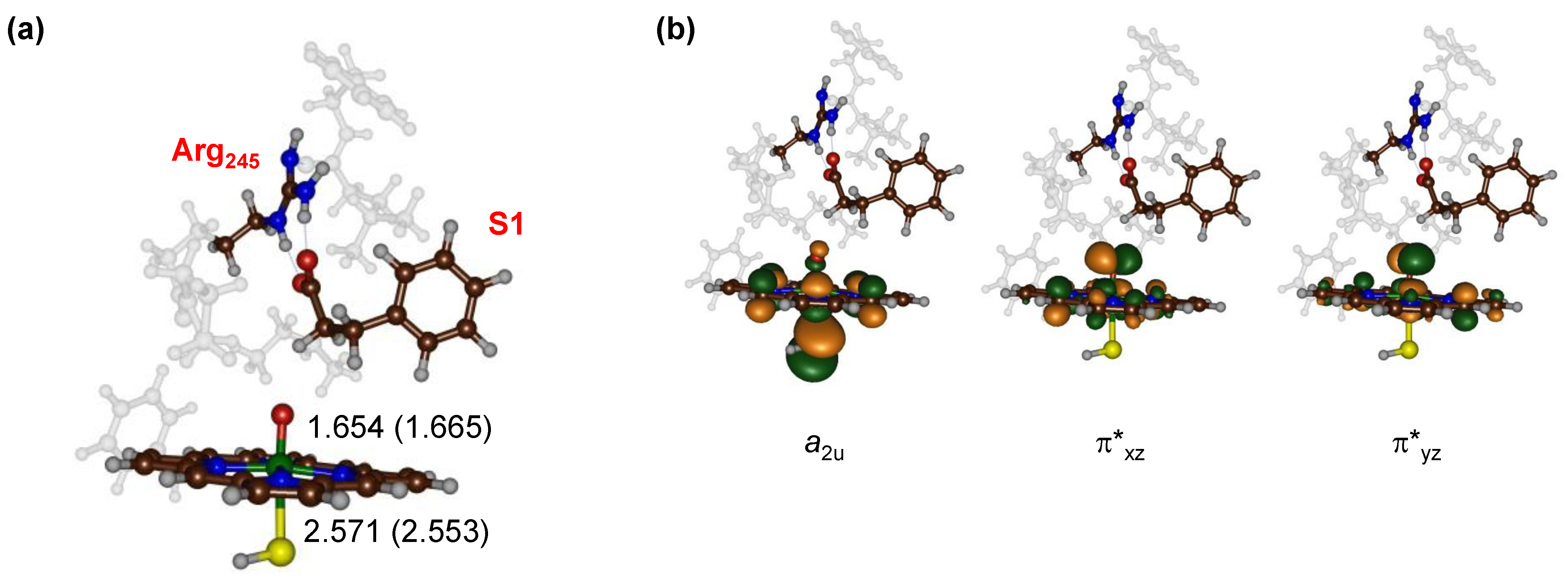
The optimized geometry of our CpdI model with substrate S1 bound (ReS1) is shown in Figure 2a for the doublet and quartet spin states. Geometrically, the doublet and quartet spin state structures are very similar with almost equal Fe–O distances of 1.665 and 1.654 Å and Fe–S interactions of 2.553 and 2.571 Å, respectively. This is not surprising as the two spin states are known to be close in energy [54,55,56,57,58,59,60] in CpdI and have the same orbital occupation with three unpaired electrons in orbitals labeled a2u, π*xz and π*yz (Figure 2b). The a2u orbital is a heme-based orbital that in D4h symmetry has contributions on the pyrrole nitrogen atoms and the meso-carbon atoms. In addition, it mixes strongly with a lone pair orbital on the sulfur atom of the axial ligand, which gives it its oxidative power. The π*xz and π*yz orbitals are the antibonding orbitals along the Fe–O bond. Similar structures and orbital occupations were obtained for the reactant complexes with substrate S2 bound (ReS2). Overall, the reactant structure and electronic configuration match previous computational studies on P450 CpdI excellently and predict similar bond lengths to those seen previously for analogous systems [36,38,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78].
Next, we explored the substrate activation channels of S1 and S2 by CpdI, and the details of the explored reaction mechanisms are shown in Scheme 3 with the designated labeling of the individual structures. Thus, structures related to substrate S1 have S1 in subscript after the label, while those associated with substrate S2 have S2 in subscript after the label. In particular, we tested the activation of the Cα–H and Cβ–H bonds of both substrates, while for S2, we also investigated the products originating from Cγ–H activation. These reaction pathways start with an initial hydrogen atom abstraction from either the Cα–H, Cβ–H or Cγ–H (if applicable) positions via transition states TS1HA,α, TS1HA,β and TS1HA,γ to form the radical intermediates designated IM1α, IM1β and IM1γ. As it is usually seen in substrate hydroxylation pathways [55,79,80], an OH rebound barrier via TS2reb,α, TS2reb,β or TS2reb,γ then gives Cα hydroxylation, Cβ hydroxylation and Cγ hydroxylation products (PrOH,α, PrOH,β and PrOH,γ), respectively. In addition, from either IM1α or IM1β a second hydrogen atom abstraction can take place via transition states TS2DS,α and TS2DS,β to form a double bond along the Cα–Cβ bond and give a desaturated product (PrDS). A final mechanism through substrate decarboxylation is only possible from IM1β via transition state TS2DC to form CO2, olefin and an iron(III)–hydroxo(heme) complex, PrDC. We investigated all these individual reaction pathways for the cluster model Re with substrates S1 and S2 bound in the quartet spin state as previous work showed the quartet spin state to be the most sensitive to bifurcation pathways [52,81].
2.1. 3-Phenylpropionate Activation by CpdI of P450 OleTJE
Next, a mechanistic study into 3-phenylpropionate (S1) activation by the CpdI model of P450 OleTJE was performed using model ReS1 leading to decarboxylation, desaturation, α-hydroxylation and β-hydroxylation products, as described above in Scheme 3. These four reaction products all originate from either Cα–H or Cβ–H hydrogen atom abstraction; hence, we will start with the hydrogen atom abstraction steps first. Figure 3 shows the hydrogen atom abstraction transition state structures TS1HA,α,S1 and TS1HA,β,S1 as geometries optimized with density functional theory methods. In addition, the middle panel of Figure 3 shows the energy profile for relative enthalpies (ΔE+ZPE; ZPE = zero-point energy). The Cβ–H hydrogen atom abstraction barrier is considerably lower in energy than the Cα–H hydrogen atom abstraction barrier: ΔE+ZPE = 14.6 kcal mol−1 for TS1HA,β,S1 and ΔE+ZPE = 20.9 kcal mol−1 for TS1HA,α,S1. Therefore, S1 activation by CpdI of P450 OleTJE will give dominant products from Cβ–H activation. The barrier for TS1HA,β,S1 is close in energy to the one calculated by a minimal cluster model without protein residues for Cβ–H activation of propane or the activation of the methyl C–H bond of trans-methyl-phenylcyclopropane [82,83,84]. The fact that the barrier TS1HA,α,S1 is much higher in energy than those barriers obtained with the minimal model and trans-methyl-phenylcyclopropane means that the second coordination sphere of the enzyme, i.e., the substrate binding pocket, restricts the substrate positioning and hampers Cα–H hydrogen atom abstraction dramatically, raising it in energy.
In general, both transition state structures are late, with long C–H bonds of 1.379 Å (TS1HA,α,S1) and 1.313 Å (TS1HA,β,S1), whereas the accepting O–H bond is much shorter: 1.176 Å for TS1HA,α,S1 and 1.261 Å for TS1HA,β,S1. In both transition states, the Fe–O distance has elongated from 1.654 in the reactant complex to 1.778 Å in TS1HA,α,S1 and 1.740 Å in TS1HA,β,S1. This is due to the formation of the O–H bond which polarizes the π*yz orbital more to the iron and hence makes the Fe–O bond weaker. Both transition states are characterized by a large imaginary frequency for the O–H–C stretch vibration, with a magnitude of i1616 cm−1 (TS1HA,α,S1) and i1670 cm−1 (TS1HA,β,S1). These large imaginary frequencies implicate that the potential energy surface is steep and gives a narrow-width peak that could result in a large kinetic isotope effect due to quantum mechanical tunneling and that will be sensitive to the substitution of hydrogen by deuterium atoms.
The transition states relax to radical intermediates IM1α,S1 and IM1β,S1 that react further past the various bifurcation channels. Let us start with the pathways from IM1α,S1 that can lead to desaturation of the Cα–Cβ bond via a second hydrogen atom abstraction or OH rebound to give the alcohol product complex; the optimized geometries and the energy landscape are shown in Figure 4. The IM1α,S1 intermediate is only 1 kcal mol−1 higher in energy than the reactant complex ReS1 and has the transition states leading to the two reaction pathways to products within 2 kcal mol−1. In particular, the lowest barrier leads to desaturation through a second hydrogen atom abstraction from Cβ–H via a transition state of ΔE+ZPE = 15.7 kcal mol−1. By contrast, the OH rebound barrier is ΔE+ZPE = 17.3 kcal mol−1. These close barriers implicate that a mixture of products may be expected for S1 activation by P450 OleTJE enzymes, which indeed is what has been observed experimentally [36]. Interestingly, the reaction leading to hydroxylation products is considerably more exothermic than the one leading to desaturation by more than 50 kcal mol−1. As such, the protein environment hampers OH rebound and affects the kinetics of the substrate activation process in favor of a much lesser exothermic pathway. This implies that P450 OleTJE reacts through negative catalysis, where an otherwise highly exothermic pathway is blocked in favor of a lesser exothermic reaction channel [85,86,87].
The transition states have typical features of OH rebound and desaturation barriers and match previous work on substrate hydroxylation and desaturation by P450 enzymes [36,38,51,52,53,64,65,66,67,68,69,72,79,88,89]. The rebound transition state has a long C–O distance of 2.354 Å, although the imaginary frequency (i469 cm−1) is clearly a pure C–O stretch vibration. As a matter of fact, a series of rebound calculations by biomimetic iron(III)–hydroxo and manganese(III)–hydroxo complexes have similarly long C–O bonds in the transition states [90,91]. In the transition state, the Fe–O bond further elongates to 1.900 Å. In the desaturation transition state TS2DS,α,S1, the same thing happens, and Fe–O elongates to 1.956 Å. Interestingly, the desaturation transition state is more reactant-like, with a shorter C–H (1.222 Å) than O–H (1.486 Å) distance.
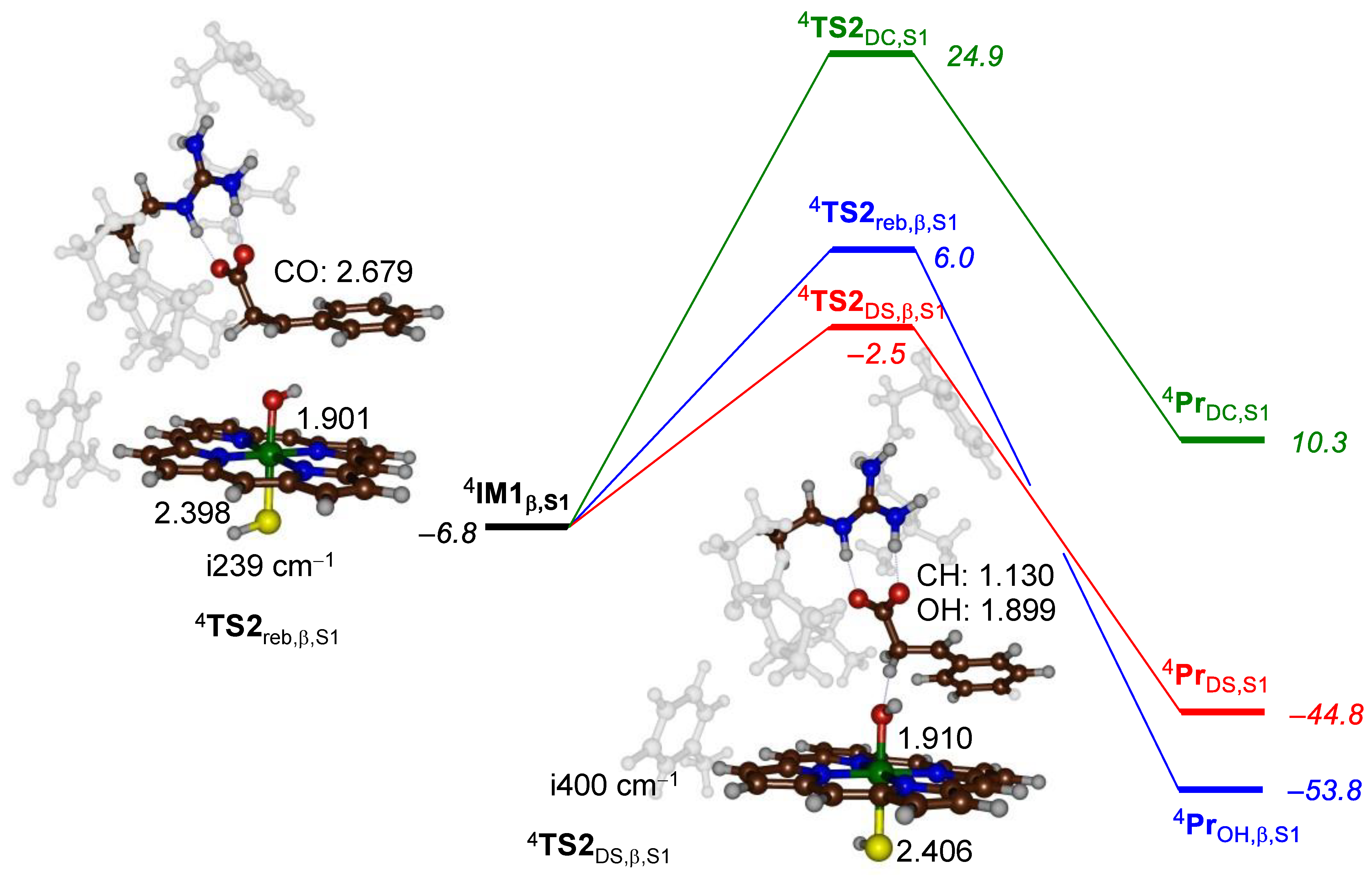
Subsequently, we investigated the pathways from the alternative radical intermediate IM1β,S1 leading to desaturation, decarboxylation and β-hydroxylation. Figure 5 shows the potential energy profile for the pathways from IM1β,S1 leading to products and the various transition states that were characterized. The lowest energy barrier (of ΔE+ZPE = 4.3 kcal mol−1 above IM1β,S1) involves a second hydrogen atom abstraction and the formation of the desaturated products. At least 8.5 kcal mol−1 above the desaturation barrier is the OH rebound barrier TS2reb,β,S1, while the decarboxylation barrier is even higher (more than 20 kcal mol−1 above the desaturation barrier).
The rebound and desaturation barriers TS2reb,β,S1 and TS2DS,β,S1 are shown in Figure 5. In both structures, the Fe–O distance is long: 1.901 and 1.910 Å for TS2reb,β,S1 and TS2DS,β,S1, respectively, which matches the structures in Figure 4 well. The rebound C–O distance is even longer in TS2reb,β,S1 than in TS2reb,α,S1 and is 2.679 Å. Nevertheless, the imaginary mode in the transition state (of i239 cm−1) represents an O–C stretch vibration; hence, it is appropriate for a rebound barrier. Similar to TS2DS,α,S1, TS2DS,β,S1 is also reactant-like, with a short C–O distance of 1.130 Å and a much longer O–H distance of 1.899 Å. Overall, these structures match previous barriers and landscapes calculated on analogous models but with different substrates.
2.2. 2-Phenylbutyrate Activation by CpdI of P450 OleTJE
To understand the effect of phenyl substitution to fatty acid substrates of P450 OleTJE, we also investigated 2-phenylbutyrate activation by our CpdI model. For this particular system, we tested hydrogen atom abstraction pathways from the Cα–H, Cβ–H and Cγ–H positions; the obtained transition states and energy landscape for these three steps are shown in Figure 6. As it can be seen, the hydrogen atom abstraction barriers 4TS1HA,α,S2 and 4TS1HA,β,S2 are within 1 kcal mol−1 in energy, although inclusion of entropy and thermal corrections appears to favor the Cα–H pathway slightly. The Cγ–H hydrogen atom abstraction barrier, on the other hand, is well higher in energy (by ΔE+ZPE = 19.1 kcal mol−1) than 4TS1HA,β,S2; consequently, this pathway is unlikely to happen, and we do not expect products originating from Cγ–H hydrogen atom abstraction.
The optimized geometries of 4TS1HA,α,S2, 4TS1HA,β,S2 and 4TS1HA,γ,S2 are presented alongside the landscape in Figure 6. All three structures have their Fe–S distances in the window from 2.393 to 2.494 Å, while the Fe–O distance ranges from 1.725 for 4TS1HA,α,S2 to 1.790 Å for 4TS1HA,β,S2. All three transition states are product-like, with shorter O–H than C–H distances. Moreover, they are all characterized with a large imaginary frequency that implies a significant amount of tunneling will take place and probably the kinetics will be affected by isotopic substitution of hydrogen atoms by deuterium in the substrate.
Next, we investigated the bifurcation pathways from each of the radical intermediates 4IM1α,S2, 4IM1β,S2 and 4IM1γ,S2. The energy landscape for the product formation step from 4IM1α,S2 is shown in Figure 7, and two possibilities were considered, namely, OH rebound to form the Cα alcohol or desaturation of the Cα–Cβ bond. The lowest energy pathway is the rebound step with a barrier of about 8.6 kcal mol−1 above 4IM1α,S2. This is a significant barrier as usually rebound barriers are much smaller in energy and typically have a magnitude between 0 and 4 kcal mol−1 [79,91]. Clearly, the protein environment and hydrogen bonding interactions restrict the OH rebound and hamper and slow this process down. Nevertheless, our prediction that the products originating from 4IM1α,S2 should give α-hydroxylation matches experimental observations excellently as those were the dominant products obtained for this substrate in reaction with P450 OleTJE [36].
The rebound transition state is similar in structure to the one shown above in Figure 4, with a long Cα–O distance of 2.469 Å and a small imaginary frequency of i255 cm−1 representing the Cα–O stretch vibration. In agreement with the rebound transition states mentioned above, the Fe–O distance has elongated and is 1.898 Å in 4TS2reb,α,S2, whereas the Fe–S distance is 2.433 Å. We also attempted to locate the transition state for the second hydrogen atom abstraction from 4IM1α,S2; however, the barrier is relatively high in energy, i.e., well above that for 4TS2reb,α,S2, and an estimate from the geometry scan located it 15.8 kcal mol−1 above the radical intermediate. Consequently, we do not expect desaturation products to appear after Cα–H hydrogen atom abstraction, and this channel will give dominant Cα hydroxylation products only.
The energy landscapes for the bifurcation pathways from 4IM1β,S2 are displayed in Figure 8. The lowest energy pathway is the second hydrogen atom abstraction leading to desaturation of the Cα–Cβ bond, with a barrier of ΔE+ZPE = 3.1 kcal mol−1 above the radical intermediate. The alternative decarboxylation mechanism, however, is high in energy and well higher by 15.3 kcal mol−1 and, consequently, will have a very minor role of importance for this substrate. Indeed, experimental studies on this particular system did not record any product originating from decarboxylation of the substrate, which is consistent with the results shown here. Optimized geometries of the two bifurcation transition states 4TS2DS,β,S2 and 4TS2DC,β,S2 are also shown in Figure 8. In both cases, the Fe–O distance has elongated with respect to the reactant complex, and it is 1.916 Å for 4TS2DS,β,S2 and 1.803 Å for 4TS2DC,β,S2. The carboxylation takes place at a long C–Cα distance of 2.285 Å but has a clear C–C stretch vibration as an imaginary frequency with a magnitude of i291 cm−1. A similar size imaginary frequency of i330 cm−1 for the desaturation barrier was recorded. This value is very small for a hydrogen atom abstraction transition state and well lower than the TS1 transition states shown above in Figure 3 and Figure 6. Consequently, the second hydrogen atom abstraction will not be affected from the same major kinetic isotope effects as the TS1 barriers reported above and limited quantum mechanical tunneling will incur during the TS2DS transition. The desaturation transition state is highly reactant-like and early, with a short C–H distance of 1.163 Å and a very long O–H distance of 1.772 Å.
Overall, the calculations performed on S2 activation by our P450 OleTJE models show competitive reaction channels originating from Cα–H and Cβ–H hydrogen atom abstraction. However, the former channel leads to dominant Cα hydroxylation products, whereas the latter channel gives desaturation instead. As such, the calculations predict a mixture of products. This is in good agreement with the product distributions reported by Pickl et al. [36] that gave a ratio of 332:80 for Cα hydroxylation versus desaturation of 2-phenylbutyrate by P450 OleTJE. To understand the differences in product distributions obtained for S1 and S2 substrates, we analyzed the electronic properties of the various structures and investigated C–H bond strengths of the substrates.
3. Discussion
This work studies substrate activation by P450 OleTJE enzymes. In particular, the substrates 3-phenylpropionate (S1) and 2-phenylbutyrate (S2) and the pathways leading to α-hydroxylation, β-hydroxylation, desaturation and decarboxylation were investigated. DFT cluster models of the enzyme mechanism show that the reaction is stepwise via a radical intermediate and proceeds with a rate-determining hydrogen atom abstraction step. The absolute energies of the barriers are quite different for the two substrates, whereby for S1, the lowest barrier is for Cβ–H hydrogen atom abstraction (ΔE+ZPE = 14.6 kcal mol−1), while the Cα–H barrier is well higher (ΔE+ZPE = 20.9 kcal mol−1). By contrast, for substrate S2, the two barriers are within 0.2 kcal mol−1 with a small preference of the Cβ–H channel. As such, a mixture of products is expected for S2. In particular, for substrate S2, the Cα–H hydrogen atom abstraction is followed by a low-energy OH rebound barrier to give α-hydroxylation, whereas the Cβ–H hydrogen atom abstraction leads to desaturation with a subsequent second hydrogen atom abstraction. These predicted products, i.e., α-hydroxylation and desaturation, for substrate S2 activation by P450 OleTJE are in perfect agreement with the product distributions reported by Pickl et al. [36].
To understand the difference in hydrogen atom abstraction barriers between the two substrates, we calculated C–H bond dissociation energies (BDEs) for isolated substrates in the gas phase at the B3LYP/6-311++G** level of theory (with a solvent model included). These BDEs were calculated from the adiabatic homolytic bond strengths as the difference in energy (ΔE+ZPE) of the substrate, a hydrogen atom and the substrate with a hydrogen atom removed. Previously, we showed that hydrogen atom abstraction barriers often correlate with the BDE of the substrate C–H bond that is broken [92,93,94], Figure 9. For substrate S1, the Cα–H bond has a BDECH, S1,α = 88.4 kcal mol−1, while the Cβ–H bond has a strength of 80.8 kcal mol−1. As such, the Cβ–H bond is the weakest bond in the substrate and the most likely bond to be activated by the enzyme. Indeed, our potential energy landscape (Figure 3) for S1 activation by P450 OleTJE gives a lower hydrogen atom abstraction barrier for Cβ–H than for Cα–H, in line with the differences in the bond strengths of the two bonds in the substrate. Clearly, the protein pocket does not affect the selectivity of the reaction, and P450 OleTJE appears to abstract the weakest C–H bond of the substrate.
We also calculated the Cα–H, Cβ–H and Cγ–H bond strengths of substrate S2 (see Figure 9). Not surprisingly, the weakest bond is the tertiary Cα–H bond with a BDE = 78.0 kcal mol−1. Much stronger are the Cβ–H bond (BDE = 91.0 kcal mol−1) and Cγ–H (95.8 kcal mol−1). Indeed, our DFT-calculated energy landscape for hydrogen atom abstraction from these three positions returns a high energy barrier for Cγ–H abstraction of ΔE+ZPE = 36.0 kcal mol−1, and hence we do not expect this reaction channel to occur. Interestingly, despite the fact that the BDEs for Cα–H and Cβ–H are widely apart, P450 OleTJE appears to be able to activate both positions, and we find almost equal hydrogen atom abstraction barriers for these pathways. Therefore, the second coordination sphere and substrate binding and positioning affect the chemoselectivity of the reaction and make the Cβ–H abstraction accessible. This is a form of negative catalysis [85,86,87] and means an otherwise high-energy reaction channel is stabilized by the protein environment and makes alternative products possible. In particular, the approach of the Cα–H bond of S2 on CpdI is sterically hindered through protein residues, meaning that this position is less accessible than the Cβ–H position of the substrate. The shape of the substrate binding pocket, therefore, guides the regioselectivity and the accessibility to the substrate and overrules thermochemical preferences such as the C–H bond strength.
4. Materials and Methods
We used density functional theory (DFT) methods as implemented in the Gaussian-09 software package [95]. The studies used the unrestricted hybrid density functional method B3LYP [96,97] for all calculations. The initial geometry optimizations, constraint geometry scans and analytical frequency calculations used a modest LACVP basis set on iron (with core potential) [98] coupled to 6-31G on the rest of the atoms: basis set BS1 [99]. All calculations were run with a solvent model included through the continuous polarized continuum model with a dielectric constant representing chlorobenzene (with dielectric constant ε = 5.7) [100]. The quality of the energies was improved through single point calculations with a 6-311+G* basis set on all atoms and LACV3P+ with core potential on iron: basis set BS2. Our methods have been extensively validated and benchmarked against experimental data, and these procedures were shown to reproduce experimental product distributions and rate constants well [101,102,103,104]. Full data of all results obtained is given in the Supporting Information accompanying this paper (Supplementary Materials).
5. Conclusions
In this work, a computational study into the activation of 3-phenylpropionate and 2-phenylbutyrate by Compound I of P450 OleTJE was conducted. We set up large active site cluster models of well over 150 atoms that contain the heme and its direct ligands, the substrate and key residues in the substrate binding pocket. We then studied all mechanisms leading to Cα hydroxylation, Cβ hydroxylation, Cγ hydroxylation, Cα–Cβ desaturation and decarboxylation. We showed that differences in the size and shape of the substrate give differences in orientation and, consequently, different product distributions. Thus, for 3-phenylpropionate, dominant desaturation products were produced through a rate-determining Cβ–H hydrogen atom abstraction channel. By contrast, 2-phenylbutyrate has competing hydrogen atom abstraction barriers for Cα–H and Cβ–H abstractions and therefore gives a mixture of Cα hydroxylation and desaturation pathways. These predicted products are in excellent agreement with those reported previously from experimental studies on the same enzyme. The barrier heights were rationalized by comparing them to the C–H bond strengths in the substrate, and the effect of substrate binding and the second coordination sphere was discussed.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ijms22137172/s1.
Author Contributions
Conceptualization, S.P.d.V.; data curation, Y.-T.L.; writing—original draft preparation, S.P.d.V. Both authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available in a publicly accessible repository.
Acknowledgments
The Computational Shared Facilities of the University of Manchester is acknowledged for providing computational resources for this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Beeson, W.T.; Vu, V.V.; Span, E.A.; Phillips, C.M.; Marletta, M.A. Cellulose degradation by polysaccharide monooxygenases. Ann. Rev. Biochem. 2015, 84, 923–946. [Google Scholar] [CrossRef]
- Kang, A.; Lee, T.S. Converting sugars to biofuels: Ethanol and beyond. Bioengineering 2015, 2, 184–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, P.H.; Davies, G.J. On the catalytic mechanisms of lytic polysaccharide monooxygenases. Curr. Opin. Chem. Biol. 2016, 31, 195–207. [Google Scholar] [CrossRef]
- Lup, A.N.K.; Abnisa, F.; Daud, W.M.A.W.; Aroua, M.K. A review on reaction mechanisms of metal-catalyzed deoxygenation process in bio-oil model compounds. Appl. Catal. A 2017, 541, 87–106. [Google Scholar] [CrossRef]
- Coines, J.; Raich, L.; Rovira, C. Modeling catalytic reaction mechanisms in glycoside hydrolases. Curr. Opin. Chem. Biol. 2019, 53, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, F.; Masgrau, L. Computational modeling of carbohydrate processing enzymes reactions. Curr. Opin. Chem. Biol. 2021, 61, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, A.G. Designer metalloenzymes for synthetic biology: Enzyme hybrids for catalysis. Curr. Opin. Chem. Biol. 2020, 58, 63–71. [Google Scholar] [CrossRef]
- Das, A.; Hessin, C.; Ren, Y.; Desage-El Murr, M. Biological concepts for catalysis and reactivity: Empowering bioinspiration. Chem. Soc. Rev. 2020, 49, 8840–8867. [Google Scholar] [CrossRef]
- Ghosh, A.C.; Duboc, C.; Gennari, M. Synergy between metals for small molecule activation: Enzymes and bioinspired complexes. Coord. Chem. Rev. 2021, 428, 213606. [Google Scholar] [CrossRef]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. (Ed.) Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) Handbook of Porphyrin Science; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010. [Google Scholar]
- Grogan, G. Cytochromes P450: Exploiting diversity and enabling application as biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Fasan, R. Tuning P450 enzymes as oxidation catalysts. ACS Catal. 2012, 2, 647–666. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Groves, J.T. Oxygen activation and radical transformations in heme proteins and metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schofield, C.J.; Zhang, Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr. Opin. Struc. Biol. 1999, 9, 722–731. [Google Scholar] [CrossRef]
- Solomon, E.I.; Brunold, T.C.; Davis, M.I.; Kemsley, J.N.; Lee, S.K.; Lehnert, N.; Neese, F.; Skulan, A.J.; Yang, Y.S.; Zhou, J. Geometric and electronic structure/function correlations in non-heme iron enzymes. Chem. Rev. 2000, 100, 235–349. [Google Scholar] [CrossRef] [PubMed]
- Abu-Omar, M.M.; Loaiza, A.; Hontzeas, N. Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 2005, 105, 2227–2252. [Google Scholar] [CrossRef]
- Krebs, C.; Galonić Fujimori, D.; Walsh, C.T.; Bollinger Jr, J.M. Non-heme Fe(IV)–oxo intermediates. Acc. Chem. Res. 2007, 40, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Kovaleva, E.G.; Lipscomb, J.D. Versatility of biological non-heme Fe(II) centers in oxygen activation reactions. Nat. Chem. Biol. 2008, 4, 186–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Visser, S.P.; Kumar, D. (Eds.) Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; Royal Society of Chemistry Publishing: Cambridge, UK, 2011. [Google Scholar]
- White, M.D.; Flashman, E. Catalytic strategies of the non-heme iron dependent oxygenases and their roles in plant biology. Curr. Opin. Chem. Biol. 2016, 31, 126–135. [Google Scholar] [CrossRef] [Green Version]
- De Visser, S.P. Mechanistic insight on the activity and substrate selectivity of nonheme iron dioxygenases. Chem. Rec. 2018, 18, 1501–1516. [Google Scholar] [CrossRef] [Green Version]
- Shoji, O.; Fujishiro, T.; Nakajima, H.; Kim, M.; Nagano, S.; Shiro, Y.; Watanabe, Y. Hydrogen peroxide dependent monooxygenations by tricking the substrate recognition of cytochrome P450BSβ. Angew. Chem. Int. Ed. 2007, 46, 3656–3659. [Google Scholar] [CrossRef]
- Rude, M.A.; Baron, T.S.; Brubaker, S.; Alibhai, M.; Del Cardayre, S.B.; Schirmer, A. Terminal olefin (1-alkene) biosynthesis by a novel P450 fatty acid decarboxylase from Jeotgalicoccus Species. Appl. Environ. Microbiol. 2011, 77, 1718–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, C.; Yan, J.; Zhang, W.; Guan, W.; Lu, X.; Li, S. Hydrogen peroxide-independent production of α-alkenes by OleTJE P450 fatty acid decarboxylase. Biotechnol. Biofuels 2014, 7, 28–40. [Google Scholar] [CrossRef] [Green Version]
- Shoji, O.; Watanabe, Y. Peroxygenase reactions catalyzed by cytochromes P450. J. Biol. Inorg. Chem. 2014, 19, 529–539. [Google Scholar] [CrossRef]
- Dennig, A.; Kuhn, M.; Tassoti, S.; Thiessenhusen, A.; Gilch, S.; Bülter, T.; Haas, T.; Hall, M.; Faber, K. Oxidative decarboxylation of short-chain fatty acids to 1-alkenes. Angew. Chem. Int. Ed. 2015, 54, 8819–8822. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.L.; Hsieh, C.H.; Makris, T.M. Decarboxylation of fatty acids to terminal alkenes by cytochrome P450 Compound I. J. Am. Chem. Soc. 2015, 137, 4940–4943. [Google Scholar] [CrossRef]
- Grant, J.L.; Mitchell, M.E.; Makris, T.M. Catalytic strategy for carbon–carbon bond scission by the cytochrome P450 OleT. Proc. Natl. Acad. Sci. USA 2016, 113, 10049–10054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Lan, D.; Durrani, R.; Hollmann, F. Peroxygenases en route to becoming dream catalysts. What are the opportunities and challenges? Curr. Opin. Chem. Biol. 2017, 37, 1–9. [Google Scholar] [CrossRef]
- Munro, A.W.; McLean, K.J.; Grant, J.L.; Makris, T.M. Structure and function of the cytochrome P450 peroxygenase enzymes. Biochem. Soc. Trans. 2018, 46, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Pickl, M.; Kurakin, S.; Cantú Reinhard, F.G.; Schmid, P.; Pöcheim, A.; Winkler, C.K.; Kroutil, W.; de Visser, S.P.; Faber, K. Mechanistic studies of fatty acid activation by CYP152 peroxygenases reveal unexpected desaturase activity. ACS Catal. 2019, 9, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.; Zachos, I.; Sieber, V. Production of propene from n-butanol: A three-step cascade utilizing the cytochrome P450 fatty acid decarboxylase OleTJE. ChemBioChem 2020, 21, 3273–3281. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.S.; Ali, H.S.; Faponle, A.S.; de Visser, S.P. How external perturbations affect the chemoselectivity of substrate activation by cytochrome P450 OleTJE. Phys. Chem. Chem. Phys. 2020, 22, 27178–27190. [Google Scholar] [CrossRef]
- Derat, E.; Shaik, S. The Poulos-Kraut mechanism of Compound I formation in horseradish peroxidase: A QM/MM study. J. Phys. Chem. B 2006, 110, 10526–10533. [Google Scholar] [CrossRef]
- Cho, K.-B.; Derat, E.; Shaik, S. Compound I of nitric oxide synthase: The active site protonation state. J. Am. Chem. Soc. 2007, 129, 3182–3188. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Tan, L.S. Is the bound substrate in nitric oxide synthase protonated or neutral and what is the active oxidant that performs substrate hydroxylation? J. Am. Chem. Soc. 2008, 130, 12961–12974. [Google Scholar] [CrossRef]
- Vidossich, P.; Fiorin, G.; Alfonso-Prieto, M.; Derat, E.; Shaik, S.; Rovira, C. On the role of water in peroxidase catalysis: A theoretical investigation of HRP Compound I formation. J. Phys. Chem. B 2010, 114, 5161–5169. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-X.; Postils, V.; Sun, W.; Faponle, A.S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S.P. Reactivity patterns of (protonated) Compound II and Compound I of Cytochrome P450: Which is the better oxidant? Chem. Eur. J. 2017, 23, 6406–6418. [Google Scholar] [CrossRef]
- Faponle, A.S.; Seebeck, F.P.; de Visser, S.P. Sulfoxide synthase versus cysteine dioxygenase reactivity in a nonheme iron enzyme. J. Am. Chem. Soc. 2017, 139, 9259–9270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmins, A.; Saint-André, M.; de Visser, S.P. Understanding how prolyl-4-hydroxylase structure steers a ferryl oxidant toward scission of a strong C–H bond. J. Am. Chem. Soc. 2017, 139, 9855–9866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-T.; Ali, H.S.; de Visser, S.P. Electrostatic perturbations from the protein affect C–H bond strengths of the substrate and enable negative catalysis in the TmpA biosynthesis enzyme. Chem. Eur. J. 2021, 27, 8851–8864. [Google Scholar] [CrossRef] [PubMed]
- Belcher, J.; McLean, K.J.; Matthews, S.; Woodward, L.S.; Fisher, K.; Rigby, S.E.J.; Nelson, D.R.; Potts, D.; Baynham, M.T.; Parker, D.A.; et al. Structure and biochemical properties of the alkene producing cytochrome P450 OleTJE (CYP152L1) from the Jeotgalicoccus sp. 8456 bacterium. J. Biol. Chem. 2014, 289, 6535–6550. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein databank. Nucl. Acids Res. 2000, 28, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Quesne, M.G.; Borowski, T.; de Visser, S.P. Quantum mechanics/molecular mechanics modelling of enzymatic processes: Caveats and breakthroughs. Chem. Eur. J. 2016, 22, 2562–2581. [Google Scholar] [CrossRef]
- Mubarak, M.Q.E.; Gérard, E.F.; Blanford, C.F.; Hay, S.; de Visser, S.P. How do vanadium chloroperoxidases generate hypochlorite from hydrogen peroxide and chloride? A computational study. ACS Catal. 2020, 10, 14067–14079. [Google Scholar] [CrossRef]
- Ji, L.; Faponle, A.S.; Quesne, M.G.; Sainna, M.A.; Zhang, J.; Franke, A.; Kumar, D.; van Eldik, R.; Liu, W.; de Visser, S.P. Drug metabolism by cytochrome P450 enzymes: What distinguishes the pathways leading to substrate hydroxylation over desaturation? Chem. Eur. J. 2015, 21, 9083–9092. [Google Scholar] [CrossRef]
- Faponle, A.S.; Quesne, M.G.; de Visser, S.P. Origin of the regioselective fatty acid hydroxylation versus decarboxylation by a cytochrome P450 peroxygenase: What drives the reaction to biofuel production? Chem. Eur. J. 2016, 22, 5478–5483. [Google Scholar] [CrossRef]
- Cantú Reinhard, F.G.; Lin, Y.-T.; Stańczak, A.; de Visser, S.P. Bioengineering of cytochrome P450 OleTJE: How does substrate positioning affect the product distributions? Molecules 2020, 25, 2675–2697. [Google Scholar] [CrossRef]
- Green, M.T. Evidence for sulfur-based radicals in thiolate compound I intermediates. J. Am. Chem. Soc. 1999, 121, 7939–7940. [Google Scholar] [CrossRef]
- Ogliaro, F.; Cohen, S.; de Visser, S.P.; Shaik, S. Medium polarization and hydrogen bonding effects on Compound I of cytochrome P450: What kind of a radical is it really? J. Am. Chem. Soc. 2000, 122, 12892–12893. [Google Scholar] [CrossRef]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Kaneti, J.; Shaik, S. The experimentally elusive oxidant of cytochrome P450: A theoretical “trapping” defining more closely the “real” species. ChemBioChem 2001, 2, 848–851. [Google Scholar] [CrossRef]
- de Visser, S.P.; Shaik, S.; Sharma, P.K.; Kumar, D.; Thiel, W. Active species of horseradish peroxidase (HRP) and cytochrome P450: Two electronic chameleons. J. Am. Chem. Soc. 2003, 125, 15779–15788. [Google Scholar] [CrossRef] [PubMed]
- Hersleth, H.-P.; Ryde, U.; Rydberg, P.; Görbitz, C.H.; Andersson, K.K. Structures of the high-valent metal-ion haem–oxygen intermediates in peroxidases, oxygenases and catalases. J. Inorg. Biochem. 2006, 100, 460–476. [Google Scholar] [CrossRef] [PubMed]
- Groenhof, A.R.; Ehlers, A.W.; Lammertsma, K. Proton assisted oxygen-oxygen bond splitting in cytochrome P450. J. Am. Chem. Soc. 2007, 129, 6204–6209. [Google Scholar] [CrossRef]
- Bathelt, C.M.; Zurek, J.; Mulholland, A.J.; Harvey, J.N. Electronic structure of compound I in human isoforms of cytochrome P450 from QM/MM modeling. J. Am. Chem. Soc. 2005, 127, 12900–12908. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef]
- Rydberg, P.; Sigfridsson, E.; Ryde, U. On the role of the axial ligand in heme proteins: A theoretical study. J. Biol. Inorg. Chem. 2004, 9, 203–223. [Google Scholar] [CrossRef]
- Harvey, J.N.; Bathelt, C.M.; Mulholland, A.J. QM/MM modeling of compound I active species in cytochrome P450, cytochrome c peroxidase, and ascorbate peroxidase. J. Comput. Chem. 2006, 27, 1352–1362. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Han, K.; Zhan, C.-G. Fundamental reaction pathways for cytochrome P450-catalyzed 5′-hydroxylation and N-demethylation of nicotine. J. Phys. Chem. B 2010, 114, 9023–9030. [Google Scholar] [CrossRef] [Green Version]
- Hirao, H.; Chuanprasit, P.; Cheong, Y.Y.; Wang, X. How is a metabolic intermediate formed in the mechanism-based inactivation of cytochrome P450 by using 1,1-dimethylhydrazine: Hydrogen abstraction or nitrogen oxidation? Chem. Eur. J. 2013, 19, 7361–7369. [Google Scholar] [CrossRef]
- Hirao, H.; Cheong, Z.C.; Wang, X. Pivotal role of water in terminating enzymatic function: A density functional theory study of the mechanism-based inactivation of cytochromes P450. J. Phys. Chem. B 2012, 116, 7787–7794. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Li, H. Hydrogen abstraction of camphor catalyzed by cytochrome P450cam: A QM/MM study. J. Phys. Chem. B 2016, 120, 12312–12320. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shi, J.; Liu, Y. Oxidative rearrangement mechanism of pentalenolactone F catalyzed by cytochrome P450 CYP161C2 (PntM). Inorg. Chem. 2018, 57, 8933–8941. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y. Mechanical insights into the enzymatic cleavage of double C–C bond in poly(cis-1,4-isoprene) by the latex clearing protein. Inorg. Chem. 2020, 59, 9627–9637. [Google Scholar] [CrossRef]
- Phung, Q.M.; Pierloot, K. Low-lying electromeric states in chloro-ligated iron(IV)-oxo porphyrin as a model for compound I, studied with second-order perturbation theory based on density matrix renormalization group. J. Chem. Theory Comput. 2019, 15, 3033–3043. [Google Scholar] [CrossRef] [PubMed]
- Altarsha, M.; Benighaus, T.; Kumar, D.; Thiel, W. How is the reactivity of cytochrome P450cam affected by Thr252X mutation? A QM/MM study for X = serine, valine, alanine, glycine. J. Am. Chem. Soc. 2009, 131, 4755–4763. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Han, K. Recent density functional theory model calculations of drug metabolism by cytochrome P450. Coord. Chem. Rev. 2012, 256, 1137–1150. [Google Scholar] [CrossRef]
- Sainna, M.A.; Kumar, S.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A comprehensive test set of epoxidation rate constants by iron(IV)-oxo porphyrin complexes. Chem. Sci. 2015, 6, 1516–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepp, K.P. Heme isomers substantially affect heme’s electronic structure and function. Phys. Chem. Chem. Phys. 2017, 19, 22355–22362. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; de Visser, S.P.; Shaik, S. Multistate reactivity in styrene epoxidation by Compound I of cytochrome P450: Mechanisms of products and side products formation. Chem. Eur. J. 2005, 11, 2825–2835. [Google Scholar] [CrossRef]
- Kumar, D.; Karamzadeh, B.; Sastry, G.N.; de Visser, S.P. What factors influence the rate constant of substrate epoxidation by Compound I of cytochrome P450 and analogous iron(IV)-oxo oxidants. J. Am. Chem. Soc. 2010, 132, 7656–7667. [Google Scholar] [CrossRef]
- Quesne, M.G.; Senthilnathan, D.; Singh, D.; Kumar, D.; Maldivi, P.; Sorokin, A.B.; de Visser, S.P. Origin of the enhanced reactivity of μ-nitrido-bridged diiron(IV)-oxo porphyrinoid complexes over cytochrome P450 Compound I. ACS Catal. 2016, 6, 2230–2243. [Google Scholar] [CrossRef] [Green Version]
- Colomban, C.; Tobing, A.H.; Mukherjee, G.; Sastri, C.V.; Sorokin, A.B.; de Visser, S.P. Mechanism of oxidative activation of fluorinated aromatic compounds by N-bridged diiron-phthalocyanine. What determines the reactivity? Chem. Eur. J. 2019, 25, 14320–14331. [Google Scholar] [CrossRef]
- Shaik, S.; Cohen, S.; de Visser, S.P.; Sharma, P.K.; Kumar, D.; Kozuch, S.; Ogliaro, F.; Danovich, D. The “rebound controversy”: An overview and theoretical modeling of the rebound step in C–H hydroxylation by cytochrome P450. Eur. J. Inorg. Chem. 2004, 207–226. [Google Scholar] [CrossRef]
- Ali, H.S.; Henchman, R.H.; de Visser, S.P. Lignin biodegradation by a cytochrome P450 enzyme: A computational study into syringol activation by GcoA. Chem. Eur. J. 2020, 26, 13093–13102. [Google Scholar] [CrossRef]
- de Visser, S.P.; Ogliaro, F.; Shaik, S. How does ethene inactivate cytochrome P450 en route to its epoxidation? A density functional study. Angew. Chem. Int. Ed. 2001, 40, 2871–2874. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Shaik, S. How does product isotope effect prove the operation of a two-state “rebound” mechanism in C–H hydroxylation by cytochrome P450? J. Am. Chem. Soc. 2003, 125, 13024–13025. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Sharma, P.K.; Cohen, S.; Shaik, S. Radical clock substrates, their C–H hydroxylation mechanism by cytochrome P450 and other reactivity patterns: What does theory reveal about the clocks’ behavior? J. Am. Chem. Soc. 2004, 126, 1907–1920. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Shaik, S. Oxygen economy of cytochrome P450: What is the origin of the mixed functionality as a dehydrogenase–oxidase enzyme compared with its normal function? J. Am. Chem. Soc. 2004, 126, 5072–5073. [Google Scholar] [CrossRef]
- Rétey, J. Enzymic reaction selectivity by negative catalysis or how do enzymes deal with highly reactive intermediates? Angew. Chem. Int. Ed. 1990, 29, 355–361. [Google Scholar] [CrossRef]
- Vögeli, B.; Erb, T.J. ‘Negative’ and ‘positive catalysis’: Complementary principles that shape the catalytic landscape of enzymes. Curr. Opin. Chem. Biol. 2018, 47, 94–100. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P.; Lin, Y.-T.; Ali, H.S.; Bagh, U.K.; Mukherjee, G.; Sastri, C.V. Negative catalysis or non-Bell-Evans-Polanyi reactivity by metalloenzymes: Examples from mononuclear heme and non-heme iron oxygenases. Coord. Chem. Rev. 2021, 439, 213914. [Google Scholar] [CrossRef]
- Pangia, T.M.; Yadav, V.; Gérard, E.F.; Lin, Y.-T.; de Visser, S.P.; Jameson, G.N.L.; Goldberg, D.P. Mechanistic investigation of oxygen rebound in a mononuclear nonheme iron complex. Inorg. Chem. 2019, 58, 9557–9561. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; de Visser, S.P. Oxygen atom transfer using an iron(IV)-oxo embedded in a tetracyclic N-heterocyclic carbene system: How does the reactivity compare to Cytochrome P450 Compound I? Chem. Eur. J. 2017, 23, 2935–2944. [Google Scholar] [CrossRef] [Green Version]
- Cummins, D.C.; Alvarado, J.G.; Zaragoza, J.P.T.; Mubarak, M.Q.E.; Lin, Y.-T.; de Visser, S.P.; Goldberg, D.P. Hydroxyl transfer to carbon radicals by Mn(OH) versus Fe(OH) corrole complexes. Inorg. Chem. 2020, 59, 16053–16064. [Google Scholar] [CrossRef]
- Codola, Z.; Gamba, I.; Acuña-Pares, F.; Casadevall, C.; Clemancey, M.; Latour, J.-M.; Luis, J.M.; Lloret-Fillol, J.; Costas, M. Design of iron coordination complexes as highly active homogenous water oxidation catalysts by deuteration of oxidation-sensitive sites. J. Am. Chem. Soc. 2019, 141, 323–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Visser, S.P.; Kumar, D.; Cohen, S.; Shacham, R.; Shaik, S. A predictive pattern of computed barriers for C–H hydroxylation by Compound I of cytochrome P450. J. Am. Chem. Soc. 2004, 126, 8362–8363. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P. A valence bond modeling of trends in hydrogen abstraction barriers and transition states of hydroxylation reactions catalyzed by cytochrome P450 enzymes. J. Am. Chem. Soc. 2008, 130, 10128–10140. [Google Scholar] [CrossRef] [PubMed]
- Latifi, R.; Bagherzadeh, M.; de Visser, S.P. Origin of the correlation of the rate constant of substrate hydroxylation by nonheme iron(IV)-oxo complexes with the bond-dissociation energy of the C–H bond of the substrate. Chem. Eur. J. 2009, 15, 6651–6662. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–272. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—consistent molecular orbital methods. XII. Further extensions of Gaussian—Type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Effect of the axial ligand on substrate sulfoxidation mediated by iron(IV)-oxo porphyrin cation radical oxidants. Chem. Eur. J. 2011, 17, 6196–6205. [Google Scholar] [CrossRef]
- Yang, T.; Quesne, M.G.; Neu, H.M.; Cantú Reinhard, F.G.; Goldberg, D.P.; de Visser, S.P. Singlet versus triplet reactivity in an Mn(V)-Oxo species: Testing theoretical predictions against experimental evidence. J. Am. Chem. Soc. 2016, 138, 12375–12386. [Google Scholar] [CrossRef] [Green Version]
- Cantú Reinhard, F.G.; Faponle, A.S.; de Visser, S.P. Substrate sulfoxidation by an iron(IV)-oxo complex: Benchmarking computationally calculated barrier heights to experiment. J. Phys. Chem. A 2016, 120, 9805–9814. [Google Scholar] [CrossRef] [PubMed]
- Cantú Reinhard, F.G.; Sainna, M.A.; Upadhyay, P.; Balan, G.A.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A systematic account on aromatic hydroxylation by a cytochrome P450 model Compound I: A low-pressure mass spectrometry and computational study. Chem. Eur. J. 2016, 22, 18608–18619. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Reaction mechanism for the formation of the active species in cytochrome P450 enzymes (Compound I) in monooxygenases using dioxygen (top reaction) or in peroxygenases using H2O2 (lower reaction); SubH is the substrate.
Scheme 1.
Reaction mechanism for the formation of the active species in cytochrome P450 enzymes (Compound I) in monooxygenases using dioxygen (top reaction) or in peroxygenases using H2O2 (lower reaction); SubH is the substrate.

Scheme 2.
Substrate activation of phenyl-substituted fatty acids by P450 OleTJE: (a) Activation of 2-methyl-3-phenylpropionate. (b) Activation of 2-phenylbutyrate.
Scheme 2.
Substrate activation of phenyl-substituted fatty acids by P450 OleTJE: (a) Activation of 2-methyl-3-phenylpropionate. (b) Activation of 2-phenylbutyrate.

Figure 1.
(a) Active site of the crystal structure coordinates of the substrate-bound form of P450 OleTJE from the 4L40 protein databank file with key residues highlighted; (b) cluster models of the active site of P450 OleTJE as investigated in this work with the substrates S1 and S2 shown in the inset.
Figure 1.
(a) Active site of the crystal structure coordinates of the substrate-bound form of P450 OleTJE from the 4L40 protein databank file with key residues highlighted; (b) cluster models of the active site of P450 OleTJE as investigated in this work with the substrates S1 and S2 shown in the inset.

Figure 2.
(a) Optimized geometries of the S1-bound reactant complexes 4ReS1 (2ReS1) with bond lengths in angstroms; (b) singly occupied molecular orbitals of 4,2ReS1.
Figure 2.
(a) Optimized geometries of the S1-bound reactant complexes 4ReS1 (2ReS1) with bond lengths in angstroms; (b) singly occupied molecular orbitals of 4,2ReS1.

Scheme 3.
Reaction mechanisms explored for substrate activation by P450 OleTJE with definition of the labels for the intermediates, products and transition states.
Scheme 3.
Reaction mechanisms explored for substrate activation by P450 OleTJE with definition of the labels for the intermediates, products and transition states.

Figure 3.
Hydrogen atom abstraction steps from substrate S1 by CpdI of P450 OleTJE. The middle panel shows relative energies in kcal mol−1 as obtained at the ΔEBS2+ZPEBS1 level of theory. Hydrogen atom abstraction transition state structures are shown on the side with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1.
Figure 3.
Hydrogen atom abstraction steps from substrate S1 by CpdI of P450 OleTJE. The middle panel shows relative energies in kcal mol−1 as obtained at the ΔEBS2+ZPEBS1 level of theory. Hydrogen atom abstraction transition state structures are shown on the side with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1.

Figure 4.
Substrate desaturation and hydroxylation bifurcation pathways for substrate S1 from the radical intermediate IM1α,S1 of P450 OleTJE. The middle panel gives energies relative to 4ReS1 in kcal mol−1 as obtained at the ΔE+ZPE level of theory. Transition state structures are shown on the side with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1.
Figure 4.
Substrate desaturation and hydroxylation bifurcation pathways for substrate S1 from the radical intermediate IM1α,S1 of P450 OleTJE. The middle panel gives energies relative to 4ReS1 in kcal mol−1 as obtained at the ΔE+ZPE level of theory. Transition state structures are shown on the side with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1.

Figure 5.
Substrate desaturation, decarboxylation and hydroxylation bifurcation pathways for substrate S1 from the radical intermediate IM1β,S1 of P450 OleTJE. The energy landscape gives energies relative to 4ReS1 in kcal mol−1 as obtained at the ΔE+ZPE level of theory. Transition state structures are shown with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1.
Figure 5.
Substrate desaturation, decarboxylation and hydroxylation bifurcation pathways for substrate S1 from the radical intermediate IM1β,S1 of P450 OleTJE. The energy landscape gives energies relative to 4ReS1 in kcal mol−1 as obtained at the ΔE+ZPE level of theory. Transition state structures are shown with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1.

Figure 6.
Hydrogen atom abstraction steps from substrate S2 by CpdI of P450 OleTJE. The middle panel gives relative energies in kcal mol−1 as obtained at the ΔE+ZPE level of theory. Hydrogen atom abstraction transition state structures are shown on the side with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1.
Figure 6.
Hydrogen atom abstraction steps from substrate S2 by CpdI of P450 OleTJE. The middle panel gives relative energies in kcal mol−1 as obtained at the ΔE+ZPE level of theory. Hydrogen atom abstraction transition state structures are shown on the side with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1.

Figure 7.
Substrate desaturation and hydroxylation bifurcation pathways for substrate S2 from the radical intermediate IM1α,S2 of P450 OleTJE. The right-hand side shows energies relative to 4ReS2 in kcal mol−1 as obtained at the ΔE+ZPE level of theory. The rebound transition state structure is shown on the left with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1. The desaturation transition state energy was estimated from constraint geometry scans.
Figure 7.
Substrate desaturation and hydroxylation bifurcation pathways for substrate S2 from the radical intermediate IM1α,S2 of P450 OleTJE. The right-hand side shows energies relative to 4ReS2 in kcal mol−1 as obtained at the ΔE+ZPE level of theory. The rebound transition state structure is shown on the left with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1. The desaturation transition state energy was estimated from constraint geometry scans.

Figure 8.
Substrate desaturation and decarboxylation bifurcation pathways for substrate S2 from the radical intermediate IM1β,S2 of P450 OleTJE. The energy landscape gives energies relative to 4ReS2 in kcal mol−1 as obtained at the ΔE+ZPE level of theory. Transition state structures are shown with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1.
Figure 8.
Substrate desaturation and decarboxylation bifurcation pathways for substrate S2 from the radical intermediate IM1β,S2 of P450 OleTJE. The energy landscape gives energies relative to 4ReS2 in kcal mol−1 as obtained at the ΔE+ZPE level of theory. Transition state structures are shown with bond lengths in angstroms and the imaginary frequency in the transition state in cm−1.

Figure 9.
Calculated substrate BDE values for selected C–H bonds in S1 and S2. Values are ΔE+ZPE in kcal mol−1.
Figure 9.
Calculated substrate BDE values for selected C–H bonds in S1 and S2. Values are ΔE+ZPE in kcal mol−1.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lin, Y.-T.; de Visser, S.P. Product Distributions of Cytochrome P450 OleTJE with Phenyl-Substituted Fatty Acids: A Computational Study. Int. J. Mol. Sci. 2021, 22, 7172. https://doi.org/10.3390/ijms22137172
AMA Style
Lin Y-T, de Visser SP. Product Distributions of Cytochrome P450 OleTJE with Phenyl-Substituted Fatty Acids: A Computational Study. International Journal of Molecular Sciences. 2021; 22(13):7172. https://doi.org/10.3390/ijms22137172
Chicago/Turabian StyleLin, Yen-Ting, and Sam P. de Visser. 2021. "Product Distributions of Cytochrome P450 OleTJE with Phenyl-Substituted Fatty Acids: A Computational Study" International Journal of Molecular Sciences 22, no. 13: 7172. https://doi.org/10.3390/ijms22137172
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.