1. Introduction
Branched-chain amino acids (BCAAs) represent a group of three essential proteinogenic amino acids—valine, leucine, and isoleucine (
Figure 1). The BCAAs are associated with the synthesis of proteins and energy production, where they act as substrates [
1,
2]. Their role in enhancing muscle protein synthesis and mass during exercise training [
3,
4], in the syndrome of cachexia [
5], and aging [
6,
7] has been reported. Moreover, it was demonstrated that leucine is responsible for the activation of the anabolic signaling molecule—mammalian target of rapamycin complex 1 (mTORC1) [
8]. Besides the involvement in protein metabolism, BCAAs also affect neurotransmission and glucose metabolism [
9]. In recent years, several various novel functions of BCAAs associated with the gastrointestinal tract, immune system, mitochondrial biogenesis, or oxidative stress have been described [
10]. Moreover, novel associations between BCAAs and chronic human diseases have been reported recently [
11]. This is due to an increased number of metabolomic studies and the growing implementation of metabolomics in human disease research.
Metabolomics is a scientific field, usually focused on the identification and/or determination of a broad range of metabolites (non-targeted metabolic profiling) or a limited number of specific metabolites with a high level of chemical similarity (targeted analysis) [
12]. The crucial methodologies used in metabolomics are mainly based on a combination of liquid chromatography (LC) or gas chromatography (GC) with mass spectrometry (MS). Furthermore, capillary electrophoresis (CE) hyphenated with MS represents a very attractive separation tool for metabolomic analysis [
13,
14], because many of the metabolites in biological systems are charged and the CE is a separation technique that is well-suited for such compounds. Nevertheless, in comparison with other analytical techniques, the use of CE-MS is still underestimated [
15].
CE-MS represents a very useful analytical method for amino acid analysis that can be used for the investigation of various types of samples. In comparison with other more convenient strategies, the growing interest in this analytical tool is supported by some excellent review papers [
16,
17,
18]. The CE-MS approach combines benefits such as separation of amino acids in their native form (without the need for any excessive derivatization procedure) according to their charge-to-size ratio with highly selective and sensitive detection offering unequivocal identification and quantification of the demanded analytes present in a complex biological matrix. Moreover, CE can be characterized as a method of “green analytical chemistry”, particularly considering the aqueous separation environment, very low consumption of organic solvents, and small sample size. Among other benefits are high separation efficiency, precision comparable to the LC, ease of automation, simplicity, high sample throughput due to the relatively short analysis time, low cost, and the fact that there is little or no sample pretreatment necessary [
19].
In CE, amino acids can be separated as cations or anions depending on the composition of the background electrolyte—BGE [
20]. The selection of an appropriate separation environment in the case of the CE-MS online combination is limited by the demands on volatility and low ion strength. Therefore, small molecular organic acids, such as formic (HFo) or acetic (HAc) acid, are the preferred BGE constituents. This fact is supported by some previously published papers dealing with CE-MS analysis of proteinogenic amino acids, especially in biological fluids [
21,
22,
23,
24,
25,
26,
27,
28,
29] or with CE analysis in combination with conductivity detection focused on BCAAs only [
30,
31].
The reported CE-MS strategies are typically based on a targeted metabolomic approach that covers a large number of analytes—metabolites. Except for one study, the baseline separation between isobaric compounds such as isoleucine and leucine usually was not obtained [
25]. The absence of appropriate resolution between the analytes may cause problems with the exact quantification of the compounds and, finally, it may lead to misinterpretation of the measured data. A fine-tuning of the separation conditions and working parameters is therefore necessary. Some papers have presented improvement in the resolution between the aforementioned isobaric amino acids [
23], but the baseline separation was not obtained.
The CE-MS quantification of BCAAs is usually performed in biological fluids; however, this analytical strategy could also be effectively implemented in the analysis of food supplements and pharmaceutical quality control. To the best of our knowledge, no study has been conducted and published dealing with such an advanced approach in this area. The quality control of a nutritional supplement containing BCAAs has been performed only with the use of capillary zone electrophoresis [
32] or micellar electrokinetic chromatography (MEKC) with UV detection [
33,
34]. However, the demand for accurate and highly reliable analytical methods capable of ensuring and proving the high quality of food supplements or pharmaceuticals is still a subject of interest.
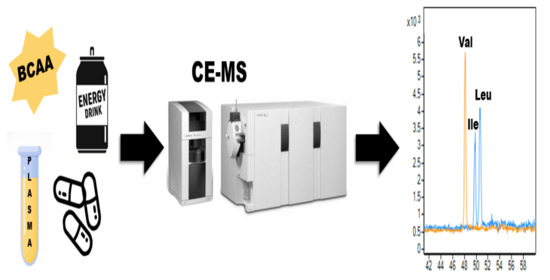
The aim of this work was the development of a simple CE-MS method for precise determination of BCAAs in various types of samples characterized by complex matrices (i.e., pharmaceutical, food, and biological samples) that could be used in quality control and bioanalytical laboratories. Different separation and working conditions were examined to achieve the appropriate resolution of the analytes from each other, the optimum selectivity, and the MS detection response. The developed analytical procedure was carried out after the validation procedure and was successfully applied for the determination of BCAAs in an energy drink, sport nutritional supplement, and human plasma. Such a broad application range unequivocally confirms the increasing importance of the CE methods in various fields of industry, medicine, and science.
3. Materials and Methods
3.1. Chemicals and Samples
The commercially available amino acid standard mixture in 0.1 M HCl, which contains L-valine, L-leucine, and L-isoleucine, was obtained from Sigma–Aldrich (Steinheim, Germany). The concentration of the demanded amino acids in the standard mixture was 2.5 mM. The LC-MS grade chemicals used for the preparation of the BGE and sheath liquid solution were purchased from Merck (Darmstadt, Germany), Sigma–Aldrich (Steinheim, Germany), and VWR International (Vienna, Austria). Ultra-pure CE (Agilent Technologies, Santa Clara, CA, USA) was used as a solvent for the electrolytes, sheath liquid, and samples. The electrolyte solutions were filtered before use through disposable 0.22 µm pore size membrane filters (Millipore, Molsheim, France) and were stored in the fridge before their use. An energy drink, Hell Active (Hell Energy, Szikszó, Hungary), and a food supplement (capsule) Amino BCAA Forte (Kompava, Nové Mesto nad Váhom, Slovakia) were obtained from a local store.
3.2. Instrumentation
The electrophoretic measurements were carried out using the Agilent 7100 CE system (Agilent Technologies, Santa Clara, CA, USA) coupled to an Agilent 6410 Series Triple Quadrupole (Agilent Technologies, Santa Clara, CA, USA) mass spectrometer using a co-axial sheath-liquid CE-MS interface. A sheath liquid composed of 5 mM NH4Ac in 50% (v/v) methanol water was delivered through an Agilent 1260 Infinity isocratic pump (Agilent Technologies, Santa Clara, CA, USA) at a flow rate of 8 µL/min. The electrospray (ESI) was operated in positive mode. The mass spectrometer settings comprise nebulizing gas (nitrogen) pressure, 10 psi; drying gas temperature, 300 °C; drying gas flow rate, 10 L/min; capillary voltage, 5000 V; and dwell time, 50 ms. An Agilent ChemStation B.04.03 software and Mass Hunter Work Station B.03.01 software (both from Agilent Technologies, Santa Clara, CA, USA) were used for CE and MS data acquisition and system control.
Uncoated fused silica capillaries with 50 µm inner diameter (id) and 120 cm total length (MicroSolv Technology Corporation, Eatontown, NJ, USA) were used in all experiments. Each new capillary was conditioned by applying a pressure flush of 950 mbar (standard internally generated pressure) for at least 30 min with 1 M NaOH, followed by ultra-pure CE water and BGE. At the end of each workday, the capillary was rinsed with 0.1 M NaOH for 10 min, ultra-pure CE water for 20 min, and BGE for 10 min and stored in BGE overnight. The samples were injected into the capillary hydrodynamically with a pressure of 50 mbar for 20 s. Before the sample injection, a short plug of 12.5% NH4OH was introduced into the capillary hydrodynamically by applying a pressure of 50 mbar for 5 s. Separation was performed at a voltage of +25 kV, which was gradually increased at the beginning of the separation from 0 to +25 kV for 18 s. During the separation procedure, a backpressure of 20 mbar was applied. To reduce the carry-over, before each injection, the capillary was re-equilibrated by applying negative voltage, −20 kV, for 30 s and flushing with BGE for 2 min.
3.3. Standard Solution and Sample Preparation
3.3.1. Standard Solutions, Calibration Solutions, and Quality Control (QC) Samples
The stock solution of analytical standards was prepared by appropriate dilution of the amino acid standards with 0.1 M HCl, obtaining 500 µM concentration levels. Working solutions (i.e., calibration and QC) were made by a proper dilution of the stock solution with ultra-pure CE water or by spiking the human plasma samples with the stock solution. Each sample was measured in six replicates.
Calibration standards were prepared by dilution with ultra-pure CE water to obtain the following concentrations: 5, 15, 30, 60, 125, 250, and 500 µM. Each point of the calibration line was measured in six replicates.
The QC samples were prepared at three concentration levels, i.e., 25 µM (low), 100 µM (medium), and 300 µM (high) by spiking the pooled plasma samples with the standard solution. A 10 µL aliquot of each QC sample was transferred to an Eppendorf tube followed by 30 µL of an appropriate organic solvent. After 20 min at laboratory temperature, the samples were centrifuged at 13,000× g for 10 min. The supernatant was then transferred to a CE vial and directly analyzed by the CE-MS method. Each sample was measured in three replicates.
3.3.2. Food Supplement Sample Preparation
Two types of food supplements were analyzed in this work. The beverage (energy drink Hell Active) sample was prepared as follows: 100 mL of the beverage was transferred to the volumetric flask and sonicated for 30 min. The solution was then filtered using Whatman filter paper No.1. From the filtrate, a 1.0 mL portion of the beverage was pipetted into a 100 mL volumetric flask and diluted with the ultra-pure CE water. The solution was then directly analyzed by the CE-MS method. The sample was measured in three replicates.
The commercial sports nutritional supplement Amino BCAA Forte (capsules) containing 192 mg Leu, 92 mg Ile, and 92 mg Val per capsule, was prepared in a very similar manner. The powder content of five capsules of this preparation was individually weighed and homogenized into a fine mixture. An amount of powder equivalent to the weight of one capsule (400 mg) was transferred to a 1000 mL volumetric flask using about 50 mL 0.1 M HCl. The solution was sonicated for 30 min. Dilution was made up to the mark with ultra-pure CE water, and the mixture was then filtered using Whatman filter paper No.1. A 1.0 mL portion of the filtrate was transferred into a 100 mL volumetric flask and diluted with ultra-pure CE water. The solution was then directly analyzed by the CE-MS method. The sample was measured in three replicates.
The whole procedure of the food and sports supplement sample preparation before the CE-MS analysis is graphically illustrated in
Figure 6a.
3.3.3. Plasma Sample Collection and Preparation
Real human plasma samples were obtained from two healthy male volunteers. Fasting blood samples were collected in the morning in test tubes containing EDTA. Plasma was obtained by centrifugation (12,000×
g, 10 min.) within 30 min of sample collection. The samples were then aliquoted and stored at −20 °C until further analysis. Before analysis, the plasma samples were left to thaw at 4 °C, and then 10 µL of each sample was transferred to an Eppendorf tube and 30 µL of acetonitrile was added to precipitate proteins. After 20 min at laboratory temperature, the samples were centrifuged at 13,000×
g for 10 min. The supernatant was then transferred to a CE vial and directly analyzed by the CE-MS method. Each sample was measured in three replicates. A simplified scheme of the plasma sample preparation for the CE-MS analysis is presented in
Figure 6b.
4. Conclusions
A CE-MS/MS method was, for the first time, developed and validated for the quantitation of BCAAs in food, sports supplements, and human plasma samples. The efficiency of the developed method was sufficient for baseline separation of the isobaric analytes—leucine and isoleucine. This effect was obtained owing to the fine-tuning of some separation and detection parameters. The benefits of the proposed method also include the fact that there is no need for derivatization of the samples and there is the possibility to analyze BCAAs in their naïve form, simple sample pretreatment (dilution—food and sport supplements samples, protein precipitation—plasma samples), favorable performance parameters, and exceptional reliability. Moreover, our work represents the first in-depth study dealing with the investigation of various factors that may affect separation efficiency, the intensity of the analytical signal, and resolution of the analytes (e.g., injection of short plug of highly basic solution before the sample plug, application of negative pressure during the analysis).
The complex validation realized in the presented study proved that the method can be effectively applied, not only in the quality control of food and sports supplements, but also in the biomedical environment. It was demonstrated that a precise analytical development led to a highly reliable CE-MS method suitable for the bioanalysis of BCAAs.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





