
Efficient Generation of Neural Stem Cells from Embryonic Stem Cells Using a Three-Dimensional Differentiation System


Abstract
:1. Introduction
2. Results
2.1. Early Neural Lineage Commitment by 3D Culture System
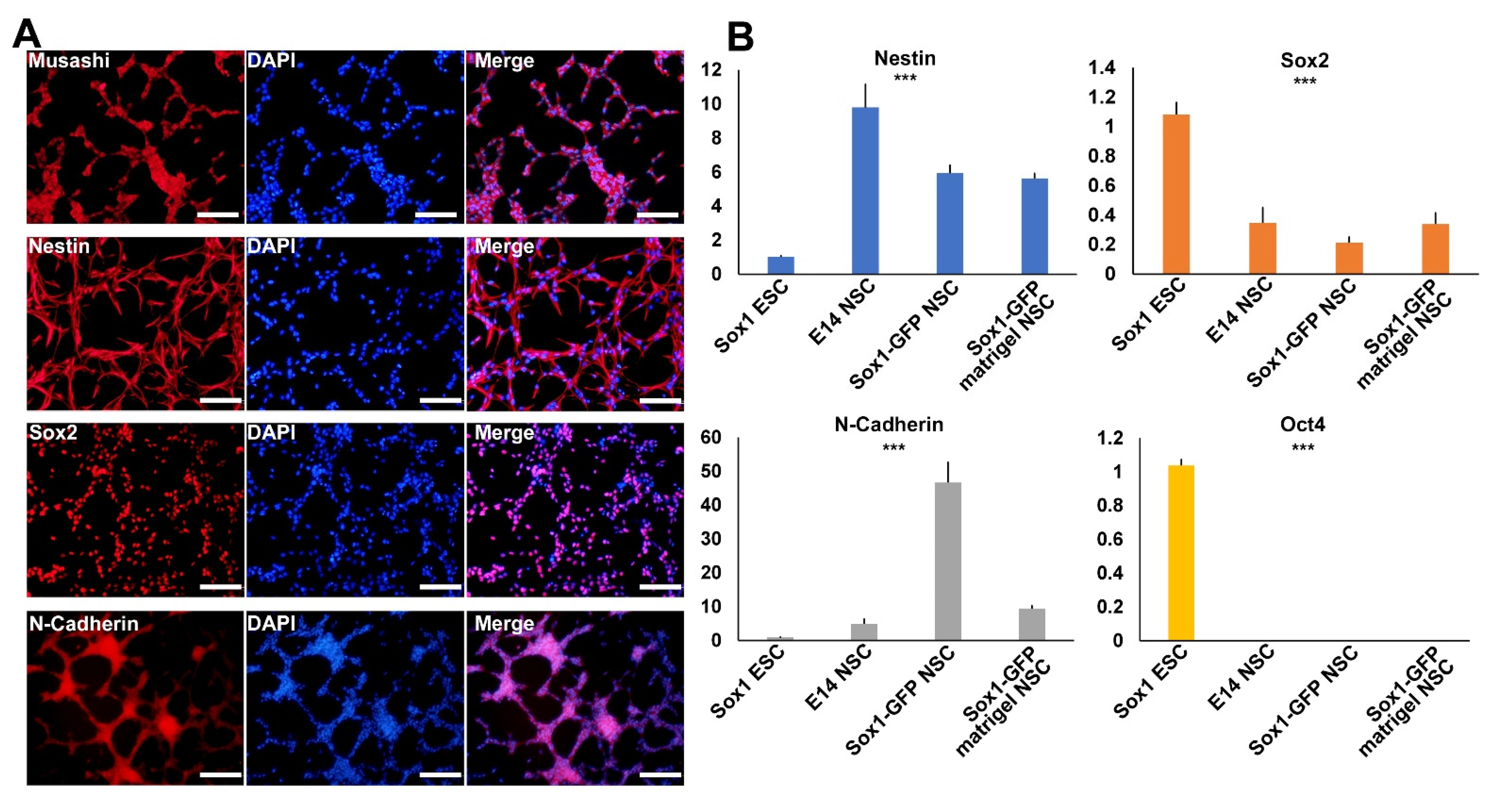
2.2. Establishment of NSCs Using Sox1-GFP-Expressing Aggregates
2.3. Characterization of Established NSCs Using NSC Markers
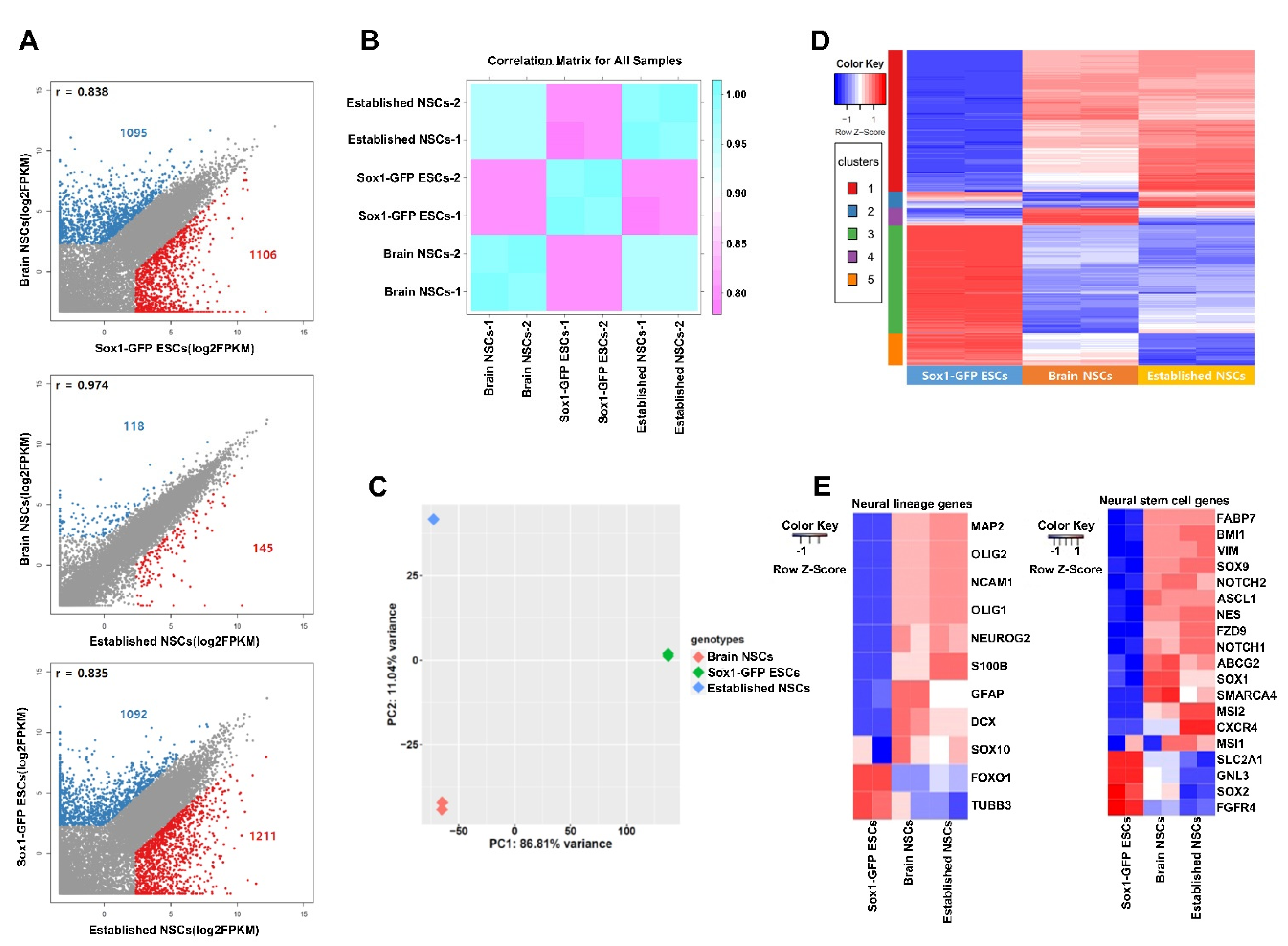
2.4. Transcriptomic Comparison of NSCs and ESCs
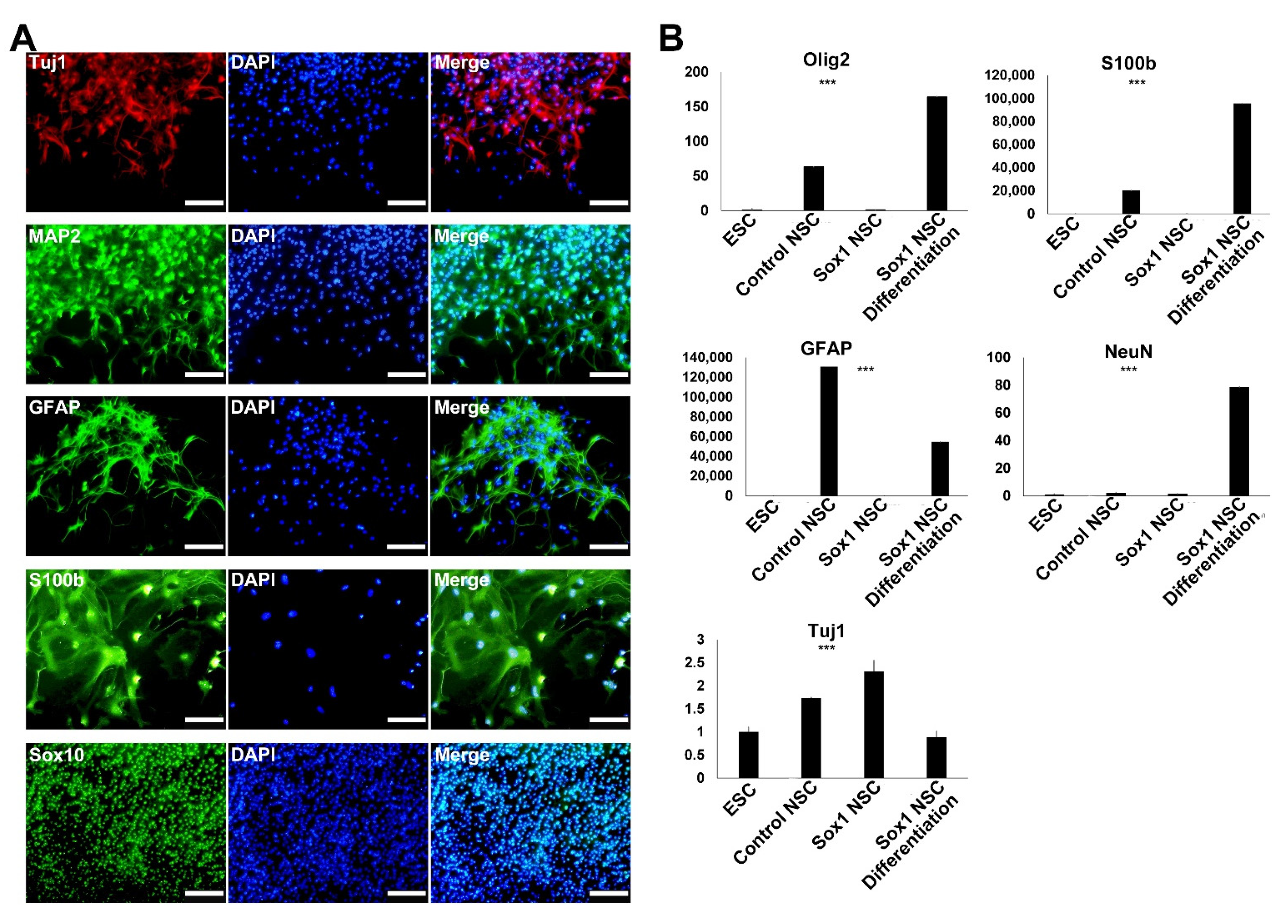
2.5. Differentiation of the Established NSCs into Neurons and Glial Cells
3. Discussion
4. Materials and Methods
4.1. Culture of Sox1-GFP ESCs
4.2. Differentiation via the 3D Matrigel Culture System
4.3. Derivation and Expansion of Neural Stem Cells (NSCs)
4.4. Differentiation of NSCs into Neurons and Glial Cells
4.5. Immunocytochemistry (ICC)
4.6. RNA Isolation and Real-Time PCR Analysis
4.7. RNA Sequencing
4.8. Sequence Read Processing
4.9. Statisitical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ying, Q.-L.; Smith, A.G. Defined conditions for neural commitment and differentiation. Methods Enzymol. 2003, 365, 327–341. [Google Scholar] [PubMed]
- Conti, L.; Pollard, S.M.; Gorba, T.; Reitano, E.; Toselli, M.; Biella, G.; Sun, Y.; Sanzone, S.; Ying, Q.-L.; Cattaneo, E. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005, 3, e283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.-R.; Ueno, Y.; Zheng, Y.-W.; Koike, N. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Kim, J.S.; Choi, S.; Hong, Y.J.; Kim, M.J.; Seo, H.G.; Do, J.T. Neural stem cells differentiated from iPS cells spontaneously regain pluripotency. Stem Cells 2014, 32, 2596–2604. [Google Scholar] [CrossRef] [PubMed]
- Bain, G.; Kitchens, D.; Yao, M.; Huettner, J.E.; Gottlieb, D.I. Embryonic stem cells express neuronal properties in vitro. Dev. Biol. 1995, 168, 342–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, H.; Mizuseki, K.; Nishikawa, S.; Kaneko, S.; Kuwana, Y.; Nakanishi, S.; Nishikawa, S.-I.; Sasai, Y. Induction of midbrain dopaminergic neurons from ES cells by stromal cell–derived inducing activity. Neuron 2000, 28, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Chambers, S.M.; Fasano, C.A.; Papapetrou, E.P.; Tomishima, M.; Sadelain, M.; Studer, L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009, 27, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.J.; Kim, J.S.; Choi, H.W.; Song, H.; Park, C.; Do, J.T. In vivo generation of neural stem cells through teratoma formation. Stem Cells Dev. 2016, 25, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Hong, Y.J.; Choi, H.W.; Song, H.; Byun, S.J.; Do, J.T. Generation of in vivo neural stem cells using partially reprogrammed cells defective in in vitro differentiation potential. Oncotarget 2017, 8, 16456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubert, J.; Stavridis, M.P.; Tweedie, S.; O’Reilly, M.; Vierlinger, K.; Li, M.; Ghazal, P.; Pratt, T.; Mason, J.O.; Roy, D.; et al. Screening for mammalian neural genes via fluorescence-activated cell sorter purification of neural precursors from Sox1-gfp knock-in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 11836–11841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, H.B.; Episkopou, V. Comparative expression of the mouse Sox1, Sox2 and Sox3 genes from pre-gastrulation to early somite stages. Mech. Dev. 1999, 86, 197–201. [Google Scholar] [CrossRef]
- Kan, L.; Jalali, A.; Zhao, L.R.; Zhou, X.; McGuire, T.; Kazanis, I.; Episkopou, V.; Bassuk, A.G.; Kessler, J.A. Dual function of Sox1 in telencephalic progenitor cells. Dev. Biol. 2007, 310, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsui, K.; Tokuzawa, Y.; Itoh, H.; Segawa, K.; Murakami, M.; Takahashi, K.; Maruyama, M.; Maeda, M.; Yamanaka, S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 2003, 113, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Voss, A.K.; Krebs, D.L.; Thomas, T. C3G regulates the size of the cerebral cortex neural precursor population. EMBO J. 2006, 25, 3652–3663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement membrane matrix with biological activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Hong, Y.J.; Kim, J.S.; Song, H.; Cho, S.G.; Bae, H.; Kim, C.; Byun, S.J.; Do, J.T. In vivo differentiation of induced pluripotent stem cells into neural stem cells by chimera formation. PLoS ONE 2017, 12, e0170735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, H.H.; Ambrosio, F.; Trumbower, R.D.; Reier, P.J.; Behrman, A.L.; Wolf, S.L. Neural stem cell therapy and rehabilitation in the central nervous system: Emerging partnerships. Phys. Ther. 2016, 96, 734–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, H.W.; Joo, J.Y.; Hong, Y.J.; Kim, J.S.; Song, H.; Lee, J.W.; Wu, G.; Schöler, H.R.; Do, J.T. Distinct enhancer activity of Oct4 in naive and primed mouse pluripotency. Stem Cell Rep. 2016, 7, 911–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | |
|---|---|---|
| Nestin | 5′-AAGTTCCCAGGCTTCTCTTG-3′ | 5′-GTCTCAAGGGTATTAGGCAAGG-3′ |
| Sox2 | 5′-GAACGCCTTCATGGTATGGTCC-3′ | 5′-CGGGTGCTCCTTCATGTGC-3′ |
| N-cadherin | 5′-GTACCGCAGCATTCCATTCAGG-3′ | 5′-GGCAATCAAGTGGAGAACCCC-3′ |
| Oct4 | 5′-GGCTTCAGACTTCGCCTTCT-3′ | 5′-TGGAAGCTTAGCCAGGTTCG-3′ |
| Olig2 | 5′-GCCTGCTCAAGTCACCGTCG-3′ | 5′-CACACGCCATCAACACTGGTCGC-3′ |
| S100b | 5′-GGTTGCCCTCATTGATGTCTTCC-3′ | 5′-CATCCCCATCTTCGTCCAGCC-3′ |
| GFAP | 5′-CTGATGTCTACCAGGCGGAGC-3′ | 5′-CCAGGTTGTTCTCTGCCTCCAG-3′ |
| NeuN | ||
| Tuj1 | 5′-GCTCACGCAGCAGATGTTCG-3′ | 5′-GGATGTCACACACGGCTACC-3′ |
| Actb | 5′-CGCCATGGATGACGATATCG-3′ | 5′-CGAAGCCGGCTTTGCACATG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, S.-H.; Bae, M.-R.; La, H.; Song, H.; Hong, K.; Do, J.-T. Efficient Generation of Neural Stem Cells from Embryonic Stem Cells Using a Three-Dimensional Differentiation System. Int. J. Mol. Sci. 2021, 22, 8322. https://doi.org/10.3390/ijms22158322
Yoon S-H, Bae M-R, La H, Song H, Hong K, Do J-T. Efficient Generation of Neural Stem Cells from Embryonic Stem Cells Using a Three-Dimensional Differentiation System. International Journal of Molecular Sciences. 2021; 22(15):8322. https://doi.org/10.3390/ijms22158322
Chicago/Turabian StyleYoon, Sang-Hoon, Mi-Rae Bae, Hyeonwoo La, Hyuk Song, Kwonho Hong, and Jeong-Tae Do. 2021. "Efficient Generation of Neural Stem Cells from Embryonic Stem Cells Using a Three-Dimensional Differentiation System" International Journal of Molecular Sciences 22, no. 15: 8322. https://doi.org/10.3390/ijms22158322