
Contribution of Ghrelin to the Pathogenesis of Growth Hormone Deficiency

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Physiology of the GH/IGF-1 Axis
3. Remarks on the Certain Limitations of GHD Diagnostics in the Transition Period from Childhood to Adult Age
- (1)
- GHD in children is treated if the GHmax value in stimulation tests indicates not only severe GHD but also the so-called partial GHD (GHmax between 5 and 10 ng/mL) [49,50], while GHD in adults includes only cases with severe GHD (GHmax < 3 ng/mL according to the majority of authors). There are discussions on whether children with partial GHD should actually be treated with rhGH, as there are reports that they do not need such a therapy [52,53].
- (2)
- (3)
- There are limitations regarding the use of stimulation tests in adults (e.g., contraindications to the stimulation test after intravenous insulin administration in patients with ischemic heart disease, epilepsy, or in the elderly) [41,42] and differences in the obtained GH stimulation results, depending on the patient’s body mass index (BMI) [56];
- (4)
- Most substances used as agents in the tests increase the secretion of GH indirectly, by stimulating the secretion of GHRH. There are only two (2) tests directly stimulating GH, namely the GHRH test and the test with use of macimorelin, which is a non-peptide synthetic GHS-R agonist [57]. Macimorelin received FDA approval for use in the diagnosis of GHD in adults in December 2018 [43,58] and it has not yet been registered for GHD diagnostics in children;
- (5)
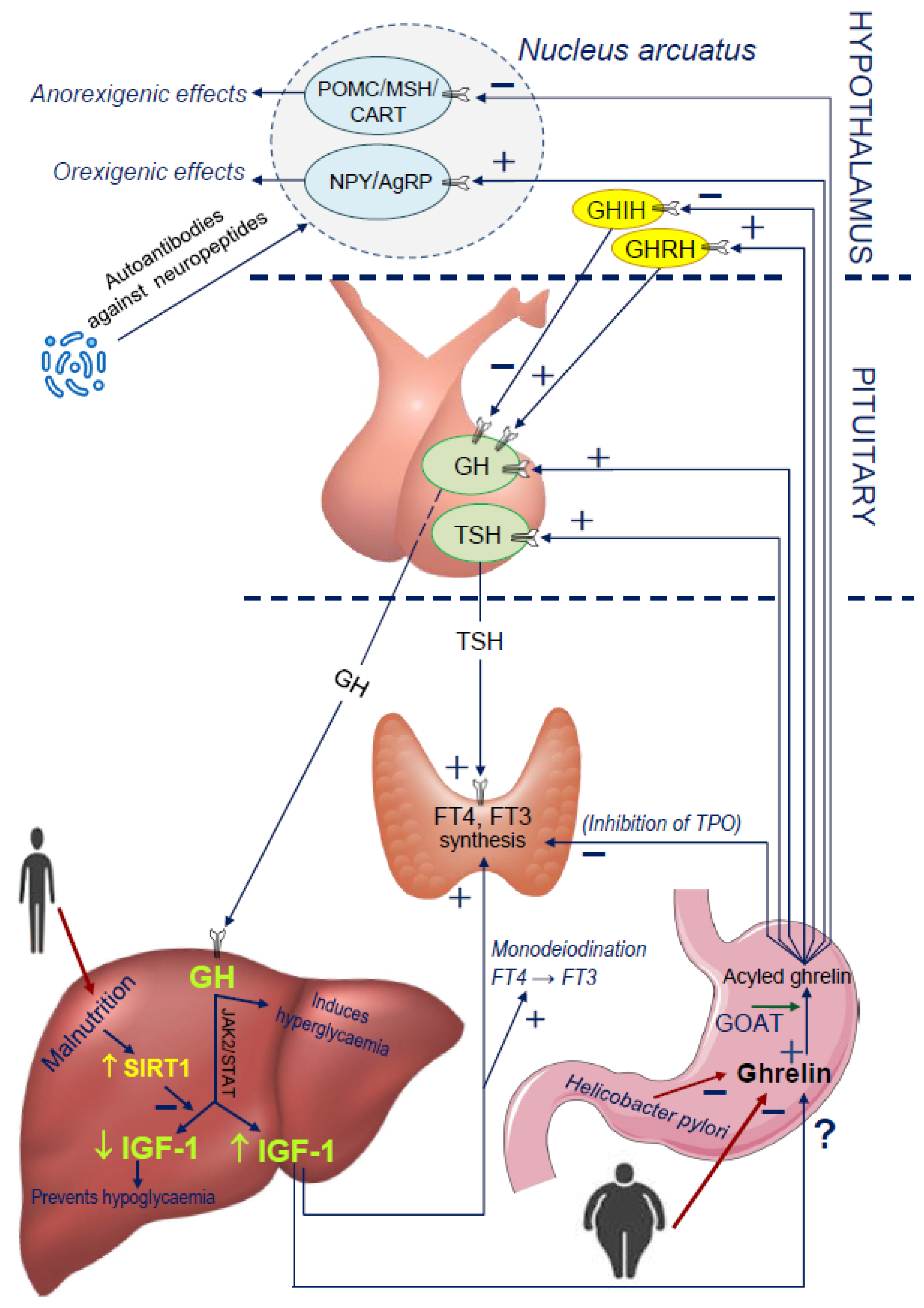
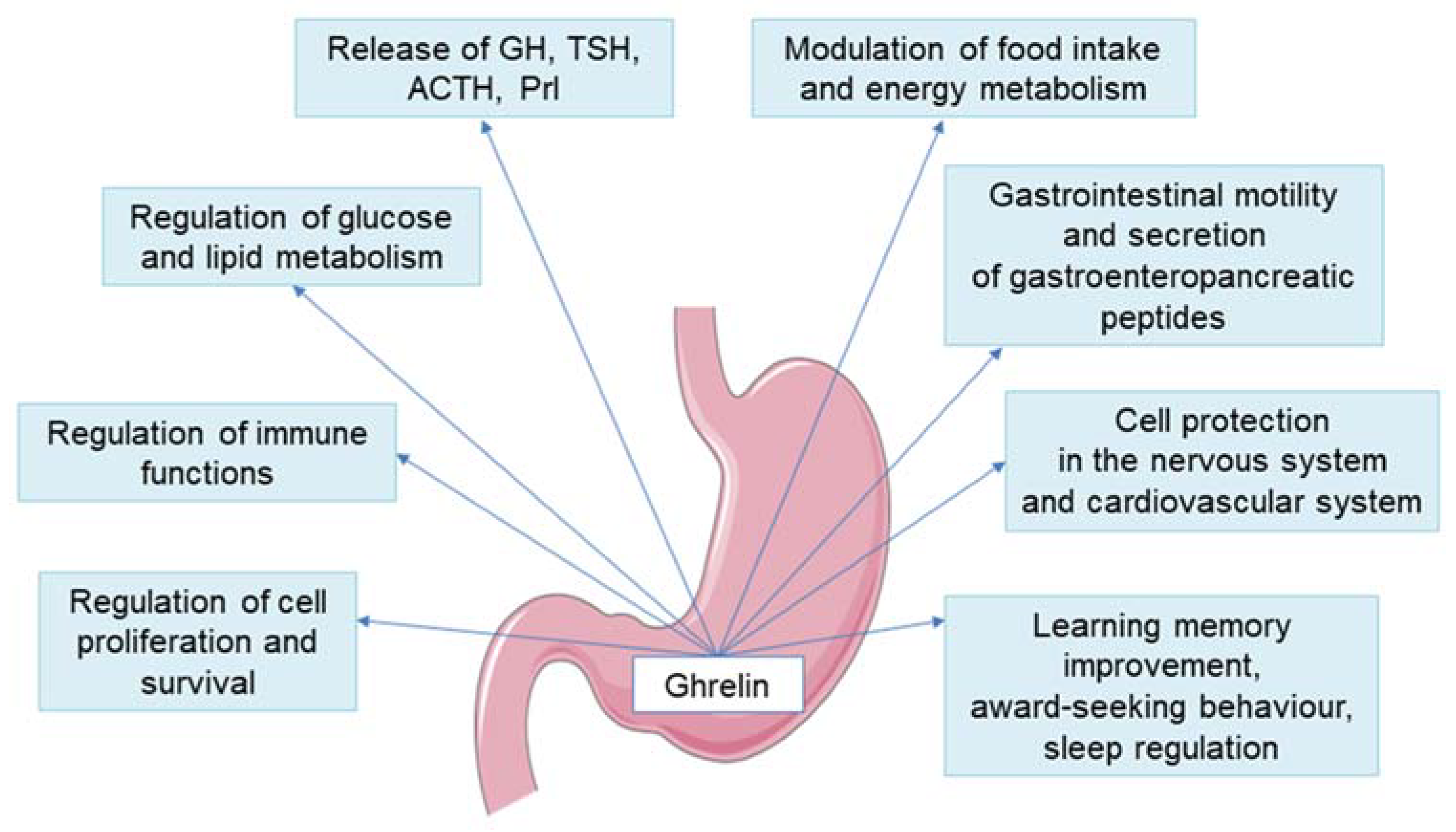
4. New Insights into the Ghrelin and GH/IGF-1 Axis Integrating System
4.1. Disorders of Nocturnal Ghrelin Secretion
4.2. GHS Receptor Mutations Leading to Short Stature and GHD
4.3. The Influence of Malnutrition on Ghrelin Secretion
4.4. Limitation in the Production of Ghrelin in the Stomach: GOAT and Helicobacter Pylori
4.5. Potential Influence of Microbiota and Molecular Mimicry on Ghrelin Secretion
4.6. Influence of TSH and FT4 on the Ghrelin-GH-IGF-1 Axis
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTH | adrenocorticotropic hormone |
| ADH | vasopressin |
| AgRP | Agouti-related peptide |
| AMPK | adenosine monophosphate-activated protein kinase |
| AO-GHD | adult-onset growth hormone deficiency |
| BMI | body mass index |
| (Ca2+)I | intracellular Ca2+ concentration |
| CART | cocaine and amphetamine regulated transcript |
| CNS | central nervous system |
| CO-GHD | childhood-onset growth hormone deficiency |
| CRH | corticotropin-releasing hormone |
| ERK | extracellular signal-regulated kinase |
| ESE | European Society of Endocrinology |
| ESPE | European Society of Pediatric Endocrinology |
| FDA | Food and Drug Administration |
| FFA | free fatty acids |
| FT3 | free triiodothyronine |
| FT4 | free thyroxine |
| GLP-1 | glucagon-like peptide 1 |
| GH | growth hormone |
| GHD | growth hormone deficiency |
| GHIH | growth hormone inhibiting hormone |
| GHmax | maximal GH level during stimulation test |
| GHR | growth hormone receptor |
| GHRH | growth hormone releasing hormone |
| GHRP-6 | growth hormone-releasing peptide 6 |
| GHS-R | growth hormone secretagogue receptor |
| GHS-R1a | isoform 1a of growth hormone secretagogue receptor |
| GHS-R1b | isoform 1b of growth hormone secretagogue receptor |
| GOAT | ghrelin O-acyl transferase |
| GST | glucagon stimulation test |
| IGF-1 | insulin-like growth factor 1 |
| IGFBP-3 | insulin-like growth factor binding protein 3 |
| IRS | insulin-receptor substrates |
| ISS | idiopathic short stature |
| ITT | insulin tolerance test |
| IUGR | intrauterine growth retardation |
| JAK | Janus kinase |
| KIMS | Pharmacia and Upjohn International Metabolic Database |
| MAPK | mitogen-activated protein kinase |
| MCH | melanin-concentrating hormone |
| mRNA | messanger ribonucleic acid |
| αMSH | α-melanocyte-stimulating hormone |
| NAD | nicotinamide adenine dinucleotide |
| NPY | neuropeptide Y |
| NREM | non-rapid eye movement |
| PI3K-AKT/PKB | phosphatidylinositol 3-kinase and AKT/protein kinase B |
| PEPCK | phosphoenolpyruvate carboxykinase |
| POMC | proopiomelanocortin |
| Prl | prolactin |
| PSIS | pituitary stalk interruption syndrome |
| rhGH | recombinant human growth hormone |
| SIRT1 | sirtuin 1 |
| STAT | signal transducer and activator of transcription |
| TPO | thyroid peroxidase |
| TSH | thyroid stimulating hormone |
References
- Hilczer, M.; Smyczynska, J.; Stawerska, R.; Lewinski, A. Final height and growth hormone secretion after completion of growth hormone therapy in patients with idiopathic growth hormone deficiency and with abnormalities of the hypothalamic-pituitary region. Neuro Endocrinol. Lett. 2005, 26, 19–24. [Google Scholar] [PubMed]
- Kokoszko, A.; Karbownik, M.; Lewiński, A. Increased lipid peroxidation in growth hormone-deficient adult patients. Neuro Endocrinol. Lett. 2006, 27, 225–230. [Google Scholar] [PubMed]
- Hilczer, M.; Smyczyńska, J.; Stawerska, R.; Lewiński, A. Effects of one-year low-dose growth hormone (GH) therapy on body composition, lipid profile and carbohydrate metabolism in young adults with childhood-onset severe GH deficiency confirmed after completion of growth promotion. Endokrynol. Pol. 2008, 59, 292–300. [Google Scholar] [PubMed]
- Karbownik-Lewinska, M.; Kokoszko, A.; Lewandowski, K.C.; Shalet, S.M.; Lewinski, A. GH replacement reduces increased lipid peroxidation in GH-deficient adults. Clin. Endocrinol. 2008, 68, 957–964. [Google Scholar] [CrossRef] [PubMed]
- Karbownik-Lewinska, M.; Lewinski, A.; McKenna, S.; Kokoszko, A.; Mucha, S.; Komorowski, J.; Krzyżanowska-Świniarska, B.; Gryczyńska, M.; Sowiński, J.; Junik, R.; et al. The Polish version of the Quality of Life Assessment of Growth Hormone Deficiency in Adults (QoL-AGHDA)—Four-stage translation and validation. Endokrynol. Pol. 2008, 59, 374–384. [Google Scholar]
- Touraine, P.; D’Souza, G.A.; Kourides, I.; Abs, R.; Barclay, P.; Xie, R.; Pico, A.; Torres-Vela, E.; Ekman, B. Lipoatrophy in GH deficient patients treated with a long-acting pegylated GH. Eur. J. Endocrinol. 2009, 161, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Olczyk, J.; Kokoszko, A.; Lewiński, A.; Karbownik-Lewińska, M. Quality of life and exercise capacity in obesity and growth hormone deficiency. Neuroendocrinol. Lett. 2010, 31, 700–707. [Google Scholar]
- McKenna, S.P.; Wilburn, J.; Twiss, J.; Crawford, S.R.; Hána, V.; Karbownik-Lewinska, M.; Popovic, V.; Pura, M.; Koltowska-Häggström, M. Adaptation of the QoL-AGHDA scale for adults with growth hormone deficiency in four Slavic languages. Health Qual. Life Outcomes 2011, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Smyczyńska, J.; Stawerska, R.; Lewiński, A.; Hilczer, M. Incidence and predictors of persistent growth hormone deficiency (GHD) in patients with isolated, childhood-onset GHD. Endokrynol. Pol. 2014, 65, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Lewiński, A.; Smyczyńska, J.; Stawerska, R.; Hilczer, M.; Stasiak, M.; Bednarczuk, T.; Bolanowski, M.; Junik, R.; Ruchała, M.; Syrenicz, A.; et al. National program of severe growth hormone deficiency treatment in adults and adolescents after Completion of Growth Promoting Therapy. Endokrynol. Pol. 2018, 69, 468–524. [Google Scholar] [CrossRef] [Green Version]
- Savage, M.O.; Burren, C.P.; Rosenfeld, R.G. The continuum of growth hormone-IGF-I axis defects causing short stature: Diagnostic and therapeutic challenges. Clin. Endocrinol. 2010, 72, 721–728. [Google Scholar] [CrossRef]
- Giavoli, C.; Ferrante, E.; Profka, E.; Olgiati, L.; Bergamaschi, S.; Ronchi, C.L.; Verrua, E.; Filopanti, M.; Passeri, E.; Montefusco, L.; et al. Influence of the d3GH receptor polymorphism on the metabolic and biochemical phenotype of GH-deficient adults at baseline and during short- and long-term recombinant human GH replacement therapy. Eur. J. Endocrinol. 2010, 163, 361–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Li, C.; Li, C.; Zhang, X. Growth hormone receptor mutations related to individual dwarfism. Int. J. Mol. Sci. 2018, 19, 1433. [Google Scholar] [CrossRef] [Green Version]
- Caicedo, D.; Díaz, O.; Devesa, P.; Devesa, J. Growth Hormone (GH) and Cardiovascular System. Int. J. Mol. Sci. 2018, 19, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arosio, M.; Ronchi, C.L.; Gebbia, C.; Cappiello, V.; Beck-Peccoz, P.; Peracchi, M. Stimulatory effects of ghrelin on circulating somatostatin and pancreatic polypeptide levels. J. Clin. Endocrinol. Metab. 2003, 88, 701–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldhuis, J.D.; Bowers, C.Y. Integrating GHS into the ghrelin system. Int. J. Pept. 2010, 879503. [Google Scholar] [CrossRef] [PubMed]
- Juul, A. Serum levels of insulin-like growth factor I and its binding proteins in health and disease. Growth Horm. IGF Res. 2003, 13, 113–170. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Sakata, I.; Sakai, T. Ghrelin cells in the gastrointestinal tract. Int. J. Pept. 2010, 945056. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhao, T.J.; Goldstein, J.L.; Brown, M.S. Inhibition of ghrelin O-acyltransferase (GOAT) by octanoylated pentapeptides. Proc. Natl. Acad. Sci. USA 2008, 105, 10750–10755. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Ogden, S.B.; Shankar, K.; Varshney, S.; Zigman, J.M. A LEAP 2 conclusions? Targeting the ghrelin system to treat obesity and diabetes. Mol. Metab. 2021, 46, 101128. [Google Scholar] [CrossRef]
- Whatmore, A.J.; Hall, C.M.; Jones, J.; Westwood, M.; Clayton, P.E. Ghrelin concentrations in healthy children and adolescents. Clin. Endocrinol. 2003, 59, 649–654. [Google Scholar] [CrossRef]
- Ghizzoni, L.; Mastorakos, G.; Vottero, A.; Ziveri, M.; Ilias, I.; Bernasconi, S. Spontaneous growth hormone (GH) secretion is not directly affected by ghrelin in either short normal prepubertal children or children with GH neurosecretory dysfunction. J. Clin. Endocrinol. Metab. 2004, 89, 5488–5495. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, K.; Tasali, E.; Leproult, R.; Scherberg, N.; Van Cauter, E. Twenty-four-hour profiles of acylated and total ghrelin: Relationship with glucose levels and impact of time of day and sleep. J. Clin. Endocrinol. Metab. 2011, 96, 486–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Wang, P.; Zheng, H.; Smith, R.G. Ghrelin stimulation of growth hormone release and appetite is mediated through the growth hormone secretagogue receptor. Proc. Natl. Acad. Sci. USA 2004, 101, 4679–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Prudom, C.E.; Nass, R.; Pezzoli, S.S.; Oliveri, M.C.; Johnson, M.L.; Veldhuis, P.; Gordon, D.A.; Howard, A.D.; Witcher, D.R.; et al. Novel ghrelin assays provide evidence for independent regulation of ghrelin acylation and secretion in healthy young men. J. Clin. Endocrinol. Metab. 2008, 93, 1980–1987. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, H.; Kangawa, K. Standard sample collections for blood ghrelin measurements. Methods Enzymol. 2012, 514, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Tauber, M.; Coupaye, M.; Diene, G.; Molinas, C.; Valette, M.; Beauloye, V. Prader-Willi syndrome: A model for understanding the ghrelin system. J. Neuroendocrinol. 2019, 31, e12728. [Google Scholar] [CrossRef]
- Holst, B.; Cygankiewicz, A.; Jensen, T.H.; Ankersen, M.; Schwartz, T.W. High constitutive signaling of the ghrelin receptor—Identification of a potent inverse agonist. Mol. Endocrinol. 2003, 17, 2201–2210. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Li, Y.; Zhang, W. The growth hormone secretagogue receptor: Its intracellular signaling and regulation. Int. J. Mol. Sci. 2014, 15, 4837–4855. [Google Scholar] [CrossRef]
- Kohno, D.; Gao, H.Z.; Muroya, S.; Kikuyama, S.; Yada, T. Ghrelin directly interacts with neuropeptide-Y-containing neurons in the rat arcuate nucleus: Ca2+ signaling via protein kinase A and N-type channel-dependent mechanisms and cross-talk with leptin and orexin. Diabetes 2003, 52, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Blum, W.F.; Albertsson-Wikland, K.; Rosberg, S.; Ranke, M.B. Serum levels of insulin-like growth factor I (IGF-I) and IGF binding protein 3 reflect spontaneous growth hormone secretion. J. Clin. Endocrinol. Metab. 1993, 76, 1610–1616. [Google Scholar] [CrossRef]
- Juul, A.; Dalgaard, P.; Blum, W.F.; Bang, P.; Hall, K.; Michaelsen, K.F.; Müller, J.; Skakkebaek, N.E. Serum levels of insulin-like growth factor (IGF)-binding protein-3 (IGFBP-3) in healthy infants, children, and adolescents: The relation to IGF-I, IGF-II, IGFBP-1, IGFBP-2, age, sex, body mass index, and pubertal maturation. J. Clin. Endocrinol. Metab. 1995, 80, 2534–2542. [Google Scholar] [CrossRef]
- Ranke, M.B.; Lindberg, A.; Tanaka, T.; Camacho-Hübner, C.; Dunger, D.B.; Geffner, M.E. Baseline characteristics and gender differences in prepubertal children treated with growth hormone in Europe, USA, and Japan: 25 years’ KIGS® experience (1987–2012) and review. Horm. Res. Paediatr. 2017, 87, 30–41. [Google Scholar] [CrossRef]
- Aguiar-Oliveira, M.H.; Souza, A.H.O.; Oliveira, C.R.P.; Campos, V.C.; Oliveira-Neto, L.A.; Salvatori, R. Mechanisms in endocrinology: The multiple facets of GHRH/GH/IGF-I axis: Lessons from lifetime, untreated, isolated GH deficiency due to a GHRH receptor gene mutation. Eur. J. Endocrinol. 2017, 177, R85–R97. [Google Scholar] [CrossRef] [PubMed]
- Di Iorgi, N.; Morana, G.; Allegri, A.E.M.; Napoli, F.; Gastaldi, R.; Calcagno, A.; Patti, G.; Loche, S.; Maghnie, M. Classical and non-classical causes of GH deficiency in the paediatric age. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 705–736. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.K.Y. Consensus guidelines for the diagnosis and treatment of adults with growth hormone deficiency: Summary statement of the Growth Hormone Research Society Workshop on Adult Growth Hormone Deficiency. J. Clin. Endocrinol. Metab. 1998, 83, 379–381. [Google Scholar] [CrossRef] [Green Version]
- Meazza, C.; Gertosio, C.; Pagani, S.; Pilotta, A.; Tinelli, C.; Buzi, F.; Farello, G.; Genoni, G.; Bona, G.; Bozzola, M. Is retesting in growth hormone deficient children really useful? Minerva Endocrinol. 2017, 42, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Brabant, G.; Poll, E.M.; Jönsson, P.; Polydorou, D.; Kreitschmann-Andermahr, I. Etiology, baseline characteristics, and biochemical diagnosis of GH deficiency in the adult: Are there regional variations? Eur. J. Endocrinol. 2009, 161, S25–S31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, S.M.; Strasburger, C.J.; Mo, D.; Hartman, M.L.; Melmed, S.; Jung, H.; Blum, W.F.; Attanasio, A.F. Changing patterns of the adult growth hormone deficiency diagnosis documented in a decade-long global surveillance database. J. Clin. Endocrinol. Metab. 2009, 94, 392–399. [Google Scholar] [CrossRef] [Green Version]
- Molitch, M.E.; Clemmons, D.R.; Malozowski, S.; Merriam, G.R.; Vance, M.L. Evaluation and treatment of adult growth hormone deficiency: An Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 2011, 96, 1587–1609. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.K.Y. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: A statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur. J. Endocrinol. 2007, 157, 695–700. [Google Scholar] [CrossRef]
- Yuen, K.C.J.; Biller, B.M.K.; Radovick, S.; Carmichael, J.D.; Jasim, S.; Pantalone, K.M.; Hoffman, A.R. American Association of Clinical Endocrinologists and American College of Endocrinology guidelines for management of growth hormone deficiency in adults and patients transitioning from pediatric to adult care. Endocr. Pract. 2019, 25, 1191–1232. [Google Scholar] [CrossRef] [PubMed]
- Clayton, P.E.; Cuneo, R.C.; Juul, A.; Monson, J.P.; Shalet, S.M.; Tauber, M. Consensus statement on the management of the GH-treated adolescent in the transition to adult care. Eur. J. Endocrinol. 2005, 152, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casanueva, F.F.; Castro, A.I.; Micic, D.; Kelestimur, F.; Dieguez, C. New guidelines for the diagnosis of growth hormone deficiency in adults. Horm. Res. 2009, 71, 112–115. [Google Scholar] [CrossRef]
- Gabellieri, E.; Chiovato, L.; Lage, M.; Castro, A.I.; Casanueva, F.F. Testing growth hormone deficiency in adults. Front. Horm. Res. 2010, 38, 139–144. [Google Scholar] [CrossRef]
- Biller, B.M.; Samuels, M.H.; Zagar, A.; Cook, D.M.; Arafah, B.M.; Bonert, V.; Stavrou, S.; Kleinberg, D.L.; Chipman, J.J.; Hartman, M.L. Sensitivity and specificity of six tests for the diagnosis of adult GH deficiency. J. Clin. Endocrinol. Metab. 2002, 87, 2067–2079. [Google Scholar] [CrossRef]
- Wagner, I.V.; Paetzold, C.; Gausche, R.; Vogel, M.; Koerner, A.; Thiery, J.; Arsene, C.G.; Henrion, A.; Guettler, B.; Keller, E.; et al. Clinical evidence-based cutoff limits for GH stimulation tests in children with a backup of results with reference to mass spectrometry. Eur. J. Endocrinol. 2014, 171, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Growth Hormone Research Society. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: Summary statement of the GH Research Society. J. Clin. Endocrinol. Metab. 2000, 85, 3990–3993. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.G.; Dattani, M.T.; Clayton, P.E. Controversies in the diagnosis and management of growth hormone deficiency in childhood and adolescence. Arch. Dis. Child. 2016, 101, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Devesa, J. Clonidine plus GHRH administration for diagnosing growth hormone deficiency in children. J. Clin. Mol. Endocrinol. 2017, 2, 6. [Google Scholar] [CrossRef]
- Smyczyńska, J.; Lewiński, A.; Hilczer, M.; Stawerska, R.; Karasek, M. Partial growth hormone deficiency (GHD) in children has more similarities to idiopathic short stature than to severe GHD. Endokrynol. Pol. 2007, 58, 182–187. [Google Scholar]
- Krukowska-Andrzejczyk, B.; Kalina, M.; Kalina-Faska, B.; Małecka-Tendera, E. Growth hormone therapy in children with partial growth hormone deficiency. Are we treating the right patients? Pediatr. Endocrinol. Diabetes Metab. 2020, 26, 65–72. [Google Scholar] [CrossRef]
- Ghigo, E.; Bellone, J.; Aimaretti, G.; Bellone, S.; Loche, S.; Cappa, M.; Bartolotta, E.; Dammacco, F.; Camanni, F. Reliability of provocative tests to assess growth hormone secretory status. Study in 472 normally growing children. J. Clin. Endocrinol. Metab. 1996, 81, 3323–3327. [Google Scholar] [CrossRef] [Green Version]
- Smyczyńska, J.; Stawerska, R.; Lewiński, A.; Hilczer, M. Do IGF-I concentrations better reflect growth hormone (GH) action in children with short stature than the results of GH stimulating tests? Evidence from the simultaneous assessment of thyroid function. Thyroid Res. 2011, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Scacchi, M.; Orsini, F.; Cattaneo, A.; Grasso, A.; Filippini, B.; PecoriGiraldi, F.; Fatti, L.M.; Moro, M.; Cavagnini, F. The diagnosis of GH deficiency in obese patients: A reappraisal with GHRH plus arginine testing after pharmacological blockade of lipolysis. Eur. J. Endocrinol. 2010, 163, 201–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, T.; Lightman, S.L. A simple test for growth hormone deficiency in adults. J. Clin. Endocrinol. Metab. 2000, 85, 1473–1476. [Google Scholar] [CrossRef]
- Garcia, J.M.; Biller, B.; Korbonits, M.; Popovic, V.; Luger, A.; Strasburger, C.J.; Chanson, P.; Medic-Stojanoska, M.; Schopohl, J.; Zakrzewska, A.; et al. Macimorelin as a diagnostic test for adult GH deficiency. J. Clin. Endocrinol. Metab. 2018, 103, 3083–3093. [Google Scholar] [CrossRef] [PubMed]
- Popovic, V.; Leal, A.; Micic, D.; Koppeschaar, H.P.; Torres, E.; Paramo, C.; Obradovic, S.; Dieguez, C.; Casanueva, F.F. GH-releasing hormone and GH-releasing peptide-6 for diagnostic testing in GH-deficient adults. Lancet 2000, 356, 1137–1142. [Google Scholar] [CrossRef]
- Ishida, J.; Saitoh, M.; Ebner, N.; Springer, J.; Anker, S.D.; von Haehling, S. Growth hormone secretagogues: History, mechanism of action, and clinical development. JCSM Rapid Commun. 2020, 3, 25–37. [Google Scholar] [CrossRef]
- Sigalos, J.T.; Pastuszak, A.W. The safety and efficacy of growth hormone secretagogues. Sex. Med. Rev. 2018, 6, 45–53. [Google Scholar] [CrossRef]
- Dzaja, A.; Dalal, M.A.; Himmerich, H.; Uhr, M.; Pollmächer, T.; Schuld, A. Sleep enhances nocturnal plasma ghrelin levels in healthy subjects. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E963–E967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stawerska, R.; Smyczyńska, J.; Czkwianianc, E.; Hilczer, M.; Lewiński, A. High concentration of ghrelin in children with growth hormone deficiency and neurosecretory dysfunction. Neuroendocrinol. Lett. 2012, 33, 331–339. [Google Scholar] [PubMed]
- Stawerska, R.; Smyczyńska, J.; Czkwianianc, E.; Pisarek, H.; Hilczer, M.; Lewiński, A. Ghrelin concentration is correlated with IGF-I/IGFBP-3 molar ratio but not with GH secretion in children with short stature. Neuroendocrinol. Lett. 2012, 33, 412–418. [Google Scholar]
- Stawerska, R.; Kolasa-Kicińska, M.; Łupińska, A.; Hilczer, M.; Lewiński, A. Comparison of nocturnal and morning ghrelin concentration in children with growth hormone deficiency and with idiopathic short stature. Chronobiol. Int. 2020, 1–7. [Google Scholar] [CrossRef]
- Janssen, J.; Van Der Toorn, F.M.; Hofland, L.J.; Van Koetsveld, P.; Broglio, F.; Ghigo, E.; Lamberts, S.W.J.; van der Lely, A.J. Systemic ghrelin levels in subjects with growth hormone deficiency are not modified by one year of growth hormone replacement therapy. Eur. J. Endocrinol. 2001, 145, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Lee, W.Y.; Rhee, E.J.; Kim, S.Y.; Oh, K.W.; Yun, E.J.; Kim, S.-W. Serum ghrelin and leptin levels in adult growth hormone deficiency syndrome. Arch. Med. Res. 2006, 5, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Lango Allen, H.; Estrada, K.; Lettre, G.; Berndt, S.I.; Weedon, M.N.; Rivadeneira, F.; Willer, C.J.; Jackson, A.U.; Vedantam, S.; Raychaudhuri, S.; et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 2010, 467, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Lanktree, M.B.; Guo, Y.; Murtaza, M.; Glessner, J.T.; Bailey, S.D.; Onland-Moret, C.; Lettre, G.; Ongen, H.; Rajagopalan, R.; Johnson, T.; et al. Meta-analysis of dense genecentric association studies reveals common and uncommon variants associated with height. Am. J. Hum. Genet. 2011, 88, 6–18. [Google Scholar] [CrossRef] [Green Version]
- Casanueva, F.F.; Camiña, J.P.; Carreira, M.C.; Pazos, Y.; Varga, J.L.; Schally, A.V. Growth hormone-releasing hormone as an agonist of the ghrelin receptor GHS-R1a. Proc. Natl. Acad. Sci. USA 2008, 105, 20452–20457. [Google Scholar] [CrossRef] [Green Version]
- Pantel, J.; Legendre, M.; Cabrol, S.; Hilal, L.; Hajaji, Y.; Morisset, S.; Nivot, S.; Vie-Luton, M.-P.; Grouselle, D.; de Kerdanet, M.; et al. Loss of constitutive activity of the growth hormone secretagogue receptor in familial short stature. J. Clin. Investig. 2006, 116, 760–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugliese-Pires, P.N.; Fortin, J.P.; Arthur, T.; Latronico, A.C.; Mendonca, B.B.; Villares, S.M.F.; Arnhold, I.J.P.; Kopin, A.S.; Jorge, A.A.L. Novel inactivating mutations in the GH secretagogue receptor gene in patients with constitutional delay of growth and puberty. Eur. J. Endocrinol. 2011, 165, 233–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, H.; Kangawa, N.; Kinouchi, A.; Sakamoto, Y.; Kimura, C.; Horikawa, R.; Shigematsu, Y.; Itakura, M.; Ogata, T.; Fujieda, K. Identification and functional analysis of novel human growth hormone secretagogue receptor (GHSR) gene mutations in Japanese subjects with short stature. J. Clin. Endocrinol. Metab. 2011, 96, E373–E378. [Google Scholar] [CrossRef] [Green Version]
- Pantel, J.; Legendre, M.; Nivot, S.; Morisset, S.; Vie-Luton, M.-P.; le Bouc, Y.; Epelbaum, J.; Amselem, S. Recessive isolated growth hormone deficiency and mutations in the ghrelin receptor. J. Clin. Endocrinol. Metab. 2009, 94, 4334–4341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torz, L.J.; Osborne-Lawrence, S.; Rodriguez, J.; He, Z.; Cornejo, M.P.; Mustafá, E.R.; Jin, C.; Petersen, N.; Hedegaard, M.A.; Nybo, M.; et al. Metabolic insights from a GHSR-A203E mutant mouse model. Mol. Metab. 2020, 39, 101004. [Google Scholar] [CrossRef]
- Iwakura, H.; Kangawa, K.; Nakao, K. The regulation of circulating ghrelin—With recent updates from cell-based assays. Endocr. J. 2015, 62, 107–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, G.P.; Barreto, A.D.C.F.; Mota, M.C.; Crispim, C.A. Caloric midpoint is associated with total calorie and macronutrient intake and body mass index in undergraduate students. Chronobiol. Int. 2019, 36, 1418–1428. [Google Scholar] [CrossRef]
- Solomou, S.; Korbonits, M. The role of ghrelin in weight-regulation disorders: Implications in clinical practice. Hormones 2014, 13, 458–475. [Google Scholar] [CrossRef]
- Otto, B.; Cuntz, U.; Fruehauf, E.; Wawarta, R.; Folwaczny, C.; Riepl, R.L.; Heiman, M.L.; Lehnert, P.; Fichter, M.; Tschöp, M. Weight gain decreases elevated plasma ghrelin concentrations of patients with anorexia nervosa. Eur. J. Endocrinol. 2001, 145, 669–673. [Google Scholar] [CrossRef] [Green Version]
- Vlaardingerbroek, H.; van den Akker, E.L.T.; Hokken-Koelega, A.C.S. Appetite- and weight-inducing and -inhibiting neuroendocrine factors in Prader–Willi syndrome, Bardet–Biedl syndrome and craniopharyngioma versus anorexia nervosa. Endocr. Connect. 2021, 10, 5. [Google Scholar] [CrossRef]
- Briggs, D.I.; Enriori, P.J.; Lemus, M.B.; Cowley, M.A.; Andrews, Z.B. Diet-induced obesity causes ghrelin resistance in arcuate NPY/AgRP neurons. Endocrinology 2010, 151, 4745–4755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stawerska, R.; Czkwianianc, E.; Smyczyńska, J.; Hilczer, M.; Lewiński, A. Nutritional status in short stature children is related to both ghrelin and insulin-like growth factor I concentrations. J. Pediatr. Gastroenterol. Nutr. 2017, 64, 812–817. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Zhao, T.J.; Li, R.L.; Sherbet, D.P.; Liang, G.; Brown, M.S. Surviving starvation: Essential role of the ghrelin-growth hormone axis. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigamonti, A.E.; Pincelli, A.I.; Corrà, B.; Viarengo, R.; Bonomo, S.M.; Galimberti, D.; Scacchi, M.; Scarpini, E.; Cavagnini, F.; Müller, E.E. Plasma ghrelin concentrations in elderly subjects: Comparison with anorexic and obese patients. J. Endocrinol. 2002, 175, R1–R5. [Google Scholar] [CrossRef] [Green Version]
- Møller, N.; Jørgensen, J.O. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr. Rev. 2009, 30, 152–177. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumar, A.; Novosyadlyy, R.; Wu, Y.; Yakar, S.; LeRoith, D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm. IGF Res. 2010, 20, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, K.; Sugimoto, T.; Kaji, H.; Kanatani, M.; Kobayashi, T.; Chihara, K. Stimulatory effect of growth hormone on bone resorption and osteoclast differentiation. Endocrinology 1996, 137, 35–41. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcik, M.; Krawczyńska, A.; Antushevich, H.; Herman, A.P. Post-receptor inhibitors of the GHR-JAK2-STAT pathway in the growth hormone signal transduction. Int. J. Mol. Sci. 2018, 19, 1843. [Google Scholar] [CrossRef] [Green Version]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Ann. Rev. Pathol. Mech. Dis. 2010, 5, 253–295. [Google Scholar] [CrossRef] [Green Version]
- Bernier, M.; Paul, R.K.; Martin-Montalvo, A.; Scheibye-Knudsen, M.; Song, S.; He, H.J.; Armour, S.M.; Hubbard, B.P.; Bohr, V.A.; Wang, L.; et al. Negative regulation of STAT3 protein-mediated cellular respiration by SIRT1 protein. J. Biol. Chem. 2011, 286, 19270–19279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Iguchi, G.; Fukuoka, H.; Suda, K.; Bando, H.; Takahashi, M.; Nishizawa, H.; Seino, S.; Takahashi, Y. SIRT1 regulates adaptive response of the growth hormone-insulin-like growth factor-I axis under fasting conditions in liver. Proc. Natl. Acad. Sci. USA 2013, 110, 14948–14953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Y.; Erion, D.M.; Yuan, Z.; Dietrich, M.; Shulman, G.I.; Horvath, T.L.; Gao, Q. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 2009, 11, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwa, V. Human growth disorders associated with impaired GH action: Defects in STAT5B and JAK2. Mol. Cell. Endocrinol. 2020, 111063. [Google Scholar] [CrossRef]
- Kaplan, D.S.; Canak, A.; Isık, E.; Orkmez, M.; Kumru, B. Relationship of fibroblast growth factor 21, sirtuin 1, visfatin, and regulators in children with short stature. Growth Factors 2018, 36, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Santos, A.A.; Salvatori, R.; Nogueira, M.C.; Bueno, A.C.; Barros-Oliveira, C.S.; Leal, Â.C.G.B.; Marinho, C.G.; Damascena, N.P.; Oliveira, D.A.; Melo, M.A.; et al. Enteroendocrine Connections in Congenital Isolated GH Deficiency Due to a GHRH Receptor Gene Mutation. J. Clin. Endocrinol. Metab. 2019, 104, 2777–2784. [Google Scholar] [CrossRef] [PubMed]
- Ørgaard, A.; Holst, J.J. The role of somatostatin in GLP-1-induced inhibition of glucagon secretion in mice. Diabetologia 2017, 60, 1731–1739. [Google Scholar] [CrossRef]
- Erdmann, J.; Lippl, F.; Schusdziarra, V. Differential effect of protein and fat on plasma ghrelin levels in man. Regul. Pept. 2003, 116, 101–107. [Google Scholar] [CrossRef]
- Gokcel, A.; Gumurdulu, Y.; Kayaselcuk, F.; Serin, E.; Ozer, B.; Ozsahin, A.K.; Guvener, N. Helicobacter pylori has no effect on plasma ghrelin levels. Eur. J. Endocrinol. 2003, 148, 423–426. [Google Scholar] [CrossRef] [Green Version]
- Osawa, H.; Nakazato, M.; Date, Y.; Kita, H.; Ohnishi, H.; Ueno, H.; Shiiya, T.; Satoh, K.; Ishino, Y.; Sugano, K. Impaired production of gastric ghrelin in chronic gastritis associated with Helicobacter pylori. J. Clin. Endocrinol. Metab. 2005, 90, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Francois, F.; Roper, J.; Joseph, N.; Pei, Z.; Chhada, A.; Shak, J.R.; de Perez, A.Z.; Perez-Perez, G.I.; Blaser, M.J. The effect of H. pylori eradication on meal-associated changes in plasma ghrelin and leptin. BMC Gastroenterol. 2011, 11, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zub-Pokrowiecka, A.; Rembiasz, K.; Konturek, S.J.; Budzynski, A.; Konturek, P.C.; Budzynski, P. Ghrelin in diseases of the gastric mucosa associated with Helicobacter pylori infection. Med. Sci. Monit. 2010, 16, 493–500. [Google Scholar]
- Gao, X.Y.; Kuang, H.Y.; Liu, X.M.; Duan, P.; Yang, Y.; Ma, Z.B. Circulating ghrelin/obestatin ratio in subjects with Helicobacter pylori infection. Nutrition 2009, 25, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Nwokolo, C.U.; Freshwater, D.A.; O’Hare, P.; Randeva, H.S. Plasma ghrelin following cure of Helicobacter pylori. Gut 2003, 52, 37–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plonka, M.; Bielanski, W.; Konturek, S.J.; Targosz, A.; Sliwowski, Z.; Dobrzanska, M.; Kaminska, A.; Sito, E.; Konturek, P.C.; Brzozowski, T. Helicobacter pylori infection and serum gastrin, ghrelin and leptin in children of Polish shepherds. Dig. Liver Dis. 2006, 38, 91–97. [Google Scholar] [PubMed]
- Pacifico, L.; Anania, C.; Osborn, J.F.; Ferrara, E.; Schiavo, E.; Bonamico, M.; Chiesa, C. Long-term effects of Helicobacter pylori eradication on circulating ghrelin and leptin concentrations and body composition in prepubertal children. Eur. J. Endocrinol. 2008, 158, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Jeffery, P.L.; McGuckin, M.A.; Linden, S.K. Endocrine impact of Helicobacter pylori: Focus on ghrelin and ghrelin o-acyltransferase. World J. Gastroenterol. 2011, 17, 1249–1260. [Google Scholar] [CrossRef]
- Chmiela, M.; Gonciarz, W. Molecular mimicry in Helicobacter pylori infections. World J. Gastroenterol. 2017, 23, 3964–3977. [Google Scholar] [CrossRef]
- Fetissov, S.O.; Hamze Sinno, M.; Coëffier, M.; Bole-Feysot, C.; Ducrotté, P.; Hökfelt, T.; Déchelotte, P. Autoantibodies against appetite-regulating peptide hormones and neuropeptides: Putative modulation by gut microflora. Nutrition 2008, 24, 348–359. [Google Scholar] [CrossRef]
- Fetissov, S.O.; Hamze Sinno, M.; Coquerel, Q.; Do Rego, J.C.; Coëffier, M.; Gilbert, D.; Hökfelt, T.; Déchelotte, P. Emerging role of autoantibodies against appetite-regulating neuropeptides in eating disorders. Nutrition 2008, 24, 854–859. [Google Scholar] [CrossRef]
- Takagi, K.; Legrand, R.; Asakawa, A.; Amitani, H.; François, M.; Tennoune, N.; Coëffier, M.; Claeyssens, S.; do Rego, J.-C.; Déchelotte, P. Anti-ghrelin immunoglobulins modulate ghrelin stability and its orexigenic effect in obese mice and humans. Nat. Commun. 2013, 4, 2685. [Google Scholar] [CrossRef]
- Stawerska, R.; Czkwianianc, E.; Matusiak, A.; Smyczynska, J.; Hilczer, M.; Chmiela, M.; Lewiński, A. Prevalence of autoantibodies against some selected growth and appetite-regulating neuropeptides in serum of short children exposed to Candida albicans colonization and/or Helicobacter pylori infection: The molecular mimicry phenomenon. Neuroendocrinol. Lett. 2015, 36, 458–464. [Google Scholar] [PubMed]
- Stawerska, R.; Czkwianianc, E.; Matusiak, A.; Smyczyńska, J.; Hilczer, M.; Chmiela, M.; Lewiński, A. Assessment of ghrelin, leptin, orexin A and alpha-MSH serum concentrations and the levels of the autoantibodies against the aforementioned peptides in relation to Helicobacter pylori infections and Candida albicans colonization in children with short stature. Pediatr. Endocrinol. Diabetes Metab. 2016, 21, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Stawerska, R.; Kolasa-Kicińska, M.; Kolejwa, M.; Smyczyńska, J.; Hilczer, M.; Czkwianianc, E.; Lewiński, A. Frequency of oligosymptomatic gastrointestinal tract diseases and its relation to IGF-I in idiopathic (non-GH-deficient) short stature children. Arch. Med. Sci. 2020. [Google Scholar] [CrossRef]
- Akin, F.; Yaylali, G.F.; Turgut, S.; Kaptanoglu, B. Growth hormone/insulin-like growth factor axis in patients with subclinical thyroid dysfunction. Growth Horm. IGF Res. 2009, 19, 252–255. [Google Scholar] [CrossRef]
- Jørgensen, J.O.; Møller, J.; Laursen, T.; Orskov, H.; Christiansen, J.S.; Weeke, J. Growth hormone administration stimulates energy expenditure and extrathyroidal conversion of thyroxine to triiodothyronine in a dose-dependent manner and suppresses circadian thyrotrophin levels: Studies in GH-deficient adults. Clin. Endocrinol. 1994, 41, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Porretti, S.; Giavoli, C.; Ronchi, C.; Lombardi, G.; Zaccaria, M.; Valle, D.; Arosio, M.; Beck-Peccoz, P. Recombinant human GH replacement therapy and thyroid function in a large group of adult GH-deficient patients: When does L-T(4) therapy become mandatory? J. Clin. Endocrinol. Metab. 2002, 87, 2042–2045. [Google Scholar] [CrossRef]
- Giavoli, C.; Porretti, S.; Ferrante, E.; Cappiello, V.; Ronchi, C.L.; Travaglini, P.; Epaminonda, P.; Arosio, M.; Beck-Peccoz, P. Recombinant hGH replacement therapy and the hypothalamus-pituitary-thyroid axis in children with GH deficiency: When should we be concerned about the occurrence of central hypothyroidism? Clin. Endocrinol. 2003, 59, 806–810. [Google Scholar] [CrossRef]
- Smyczynska, J.; Hilczer, M.; Stawerska, R.; Lewinski, A. Thyroid function in children with growth hormone (GH) deficiency during the initial phase of GH replacement therapy—Clinical implications. Thyroid Res. 2010, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Reichenbach, A.; Steyn, F.J.; Sleeman, M.W.; Andrews, Z.B. Ghrelin receptor expression and colocalization with anterior pituitary hormones using a GHSR-GFP mouse line. Endocrinology 2012, 153, 5452–5466. [Google Scholar] [CrossRef]
- Rosenfeld, R.G.; Hwa, V. Biology of the somatotroph axis (after the pituitary). Ann. Endocrinol. 2017, 78, 80–82. [Google Scholar] [CrossRef]
- Gurgul, E.; Ruchała, M.; Kosowicz, J.; Zamysłowska, H.; Wrotkowska, E.; Moczko, J.; Sowiński, J. Ghrelin and obestatin in thyroid dysfunction. Endokrynol. Pol. 2012, 63, 456–462. [Google Scholar]
- Ruchala, M.; Gurgul, E.; Stangierski, A.; Wrotkowska, E.; Moczko, J. Individual plasma ghrelin changes in the same patients in hyperthyroid, hypothyroid and euthyroid state. Peptides 2014, 51, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Adamczewska, K.; Adamczewski, Z.; Łupińska, A.; Lewiński, A.; Stawerska, R. Strong positive correlation between TSH and ghrelin in euthyroid non-growth hormone-deficient children with short stature. Molecules 2020, 25, 3912. [Google Scholar] [CrossRef]
- Barington, M.; Brorson, M.M.; Hofman-Bang, J.; Rasmussen, Å.K.; Holst, B.; Feldt-Rasmussen, U. Ghrelin-mediated inhibition of the TSH-stimulated function of differentiated human thyrocytes ex vivo. PLoS ONE 2017, 12, e0184992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.J.; Hwu, C.M.; Yeh, C.C.; Wang, P.S.; Wang, S.W. Effects of subacute hypothyroidism on metabolism and growth-related molecules. Molecules 2014, 19, 1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Ghrelin | GH | IGF-1 | |
|---|---|---|---|
| Chemical structure | Protein, 28 amino acids | Protein, 191 amino acids | Protein, 70 amino acids |
| Gene location | 3p25.3 | 17q23.3 | 12q23.2 |
| Main source of synthesis and secretion | Mainly the stomach X/A-like endocrine cells in oxyntic mucosa | Only somatotropic cells | Mainly hepatocytes |
| Circadian pattern of secretion | Maintained circadian rhythm: night—stable high concentration; day—increases in the fasting state and decreases after meal | Maintained circadian rhythm: night—high peaks at III NREM phase; day—low concentration, with some peaks 3 to 5 h, lower than at night | Without circadian rhythm, day and night are similar levels—stable, half-life is over 15 h |
| Lifetime secretion model |  |  | 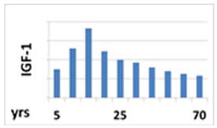 |
| Name of receptor | GH secretagogues receptor (GHS-R) | GH receptor (GHR) | Insulin-like growth factor receptor (IGF-1R) |
| Gene location for the receptor | 3q26.31 | 5p13-p12 | 15q26.3 |
| Type of receptor | Surface, seven (7) transmembrane α helix hydrophobic domains, G-protein-coupled | Surface, class I cytokine receptor | Surface, receptor with intrinsic tyrosine kinase activity |
| Main pathway important for somatotropic axis | Intracellular calcium concentrations [Ca2+]i signaling for GH secretion | JAK2-STAT for activation of IGF-1 gene | PI3K-AKT/PKB and the Ras-MAPK |
| Location of receptors | Pituitary somatotropic cells and the hypothalamus, also the stomach, heart, lungs, kidneys, intestines, adipose tissue and many other organs and tissues | GH receptors are most abundant in the liver | Ubiquitously |
| Stimulating Factor | Dosing | Time of Sample Collections (min) | Cut-Offs |
|---|---|---|---|
| Insulin | 0.1 U/kg i.v. | −30, 0, 30, 60, 90, 120 (with simultaneous serum glucose tests) | 3–5 ng/mL—Consensus, 1998 [37] 5.1 ng/mL—Biller, 2002; Molitch, 2011 [41,47] <3 ng/mL—Ho, 2007 [42] <5 ng/mL—Yuen, 2019 [43] |
| Glucagon | 1 mg i.m. (in children 30 µg/kg body weight) | 0, 90, 120, 150, 180 (glucose assessment every 30 min during the whole test) | <3 ng/mL—Ho, 2007, Yuen, 2019 [42,43] <1 ng/mL when BMI > 25kg/m2 |
| L-DOPA | 500 mg p.o. | −30, 0, 30, 60, 90 | 1.1 ng/mL; however, a test is not recommended due to the lack of adequate validation—Biller, 2002; Yuen, 2019 [43,47] |
| Arginine | 0.5 g/kg i.v. for 30 min (maximum dose 30 g) | −30, 0, 30, 60, 90, 120 | 0.4 ng/mL; however, a test is not recommended due to the lack of adequate validation—Biller, 2002; Yuen, 2019 [43,47] |
| Arginine + GHRH | 1 µg/kg i.v. bolus (GHRH), followed by a 30 min infusion of L-arginine (30 g) | −30, −15, 0, 30, 60 | 4.1 ng/mL, Biller, 2002 [47] |
| Clonidine | 0.1–0.15 mg/m2, p.o. | −30, 0, 30, 60, 90, 120 | <10 ng/mL—Wagner, 2014; Consensus, 2000; Murray, 2016 [48,49,50] |
| Clonidine + GHRH | 0.15 mg/m2, p.o. (clonidine); 1 µg/kg i.v. bolus (GHRH), at time 60 min. | −30, 0, 30, 60, 75, 90,105, 120 | <10 ng/mL—Devesa, 2017 [51] |
| Macimorelin | 0.5 mg/kg, p.o. | −30, 0, 30, 60, 90, 120, 150 | 2.8 ng/mL—according to FDA, in adults, Yuen, 2019 [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewiński, A.; Karbownik-Lewińska, M.; Wieczorek-Szukała, K.; Stasiak, M.; Stawerska, R. Contribution of Ghrelin to the Pathogenesis of Growth Hormone Deficiency. Int. J. Mol. Sci. 2021, 22, 9066. https://doi.org/10.3390/ijms22169066
Lewiński A, Karbownik-Lewińska M, Wieczorek-Szukała K, Stasiak M, Stawerska R. Contribution of Ghrelin to the Pathogenesis of Growth Hormone Deficiency. International Journal of Molecular Sciences. 2021; 22(16):9066. https://doi.org/10.3390/ijms22169066
Chicago/Turabian StyleLewiński, Andrzej, Małgorzata Karbownik-Lewińska, Katarzyna Wieczorek-Szukała, Magdalena Stasiak, and Renata Stawerska. 2021. "Contribution of Ghrelin to the Pathogenesis of Growth Hormone Deficiency" International Journal of Molecular Sciences 22, no. 16: 9066. https://doi.org/10.3390/ijms22169066