
Role of DNA-Dependent Protein Kinase in Mediating Cyst Growth in Autosomal Dominant Polycystic Kidney Disease


Abstract
:1. Introduction
2. Results
2.1. The Expression of DNA-PK Is Upregulated in Human ADPKD Transcriptome
2.2. Focal Increase of DNA-PKcs in Cyst Lining Epithelial Cells of Human ADPKD
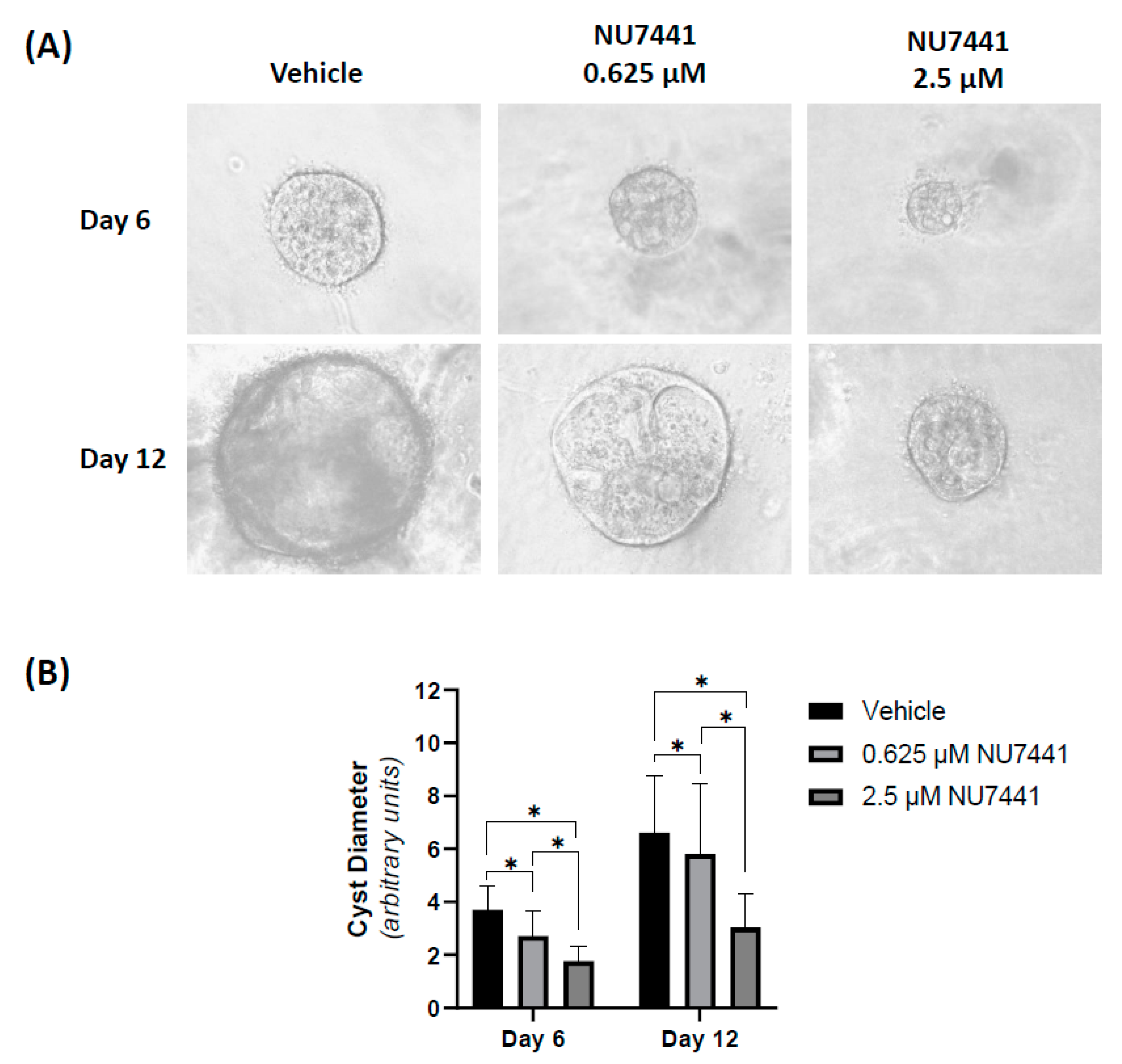
2.3. Pharmacological Inhibition of DNA-PK MDCK Cyst Growth In Vitro
2.4. DNA-PK Inhibition Does Not Cause Synthetic Lethality of Human ADPKD Cells
2.5. DNA-PK Inhibition Enhances the Anti-Proliferative Effects of Sirolimus in Both Human ADPKD and Normal Kidney Cells
3. Discussion
4. Materials and Methods
4.1. Expression of DNA-PK in Human ADPKD Transcriptome
4.2. Immunohistochemistry for DNA-PK in Human ADPKD
4.3. Cell Lines
4.4. Effect of NU7441 Treatment on Viability of ADPKD Cells
4.5. Effect of NU7441 Treatment on Three-Dimensional (3D) MDCK Cyst Model
4.6. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reeders, S.T.; Breuning, M.H.; Davies, K.E.; Nicholls, R.D.; Jarman, A.P.; Higgs, D.R.; Pearson, P.L.; Weatherall, D.J. A highly polymorphic DNA marker linked to adult polycystic kidney disease on chromosome 16. Nature 1985, 317, 542–544. [Google Scholar] [CrossRef]
- Harris, P.C.; Ward, C.J.; Peral, B.; Hughes, J. Polycystic kidney disease. 1: Identification and analysis of the primary defect. J. Am. Soc. Nephrol. 1995, 6, 1125–1133. [Google Scholar] [CrossRef]
- Mochizuki, T.; Wu, G.; Hayashi, T.; Xenophontos, S.L.; Veldhuisen, B.; Saris, J.J.; Reynolds, D.M.; Cai, Y.; Gabow, P.A.; Pierides, A.; et al. PKD2, a Gene for Polycystic Kidney Disease That Encodes an Integral Membrane Protein. Science 1996, 272, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, S.; Kubly, V.J.; Consugar, M.B.; Hopp, K.; Roy, S.; Horsley, S.W.; Chauveau, D.; Rees, L.; Barratt, T.M.; Hoff, W.G.V.; et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009, 75, 848–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopp, K.; Ward, C.J.; Hommerding, C.J.; Nasr, S.H.; Tuan, H.-F.; Gainullin, V.G.; Rossetti, S.; Torres, V.E.; Harris, P.C. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Investig. 2012, 122, 4257–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeuwen, I.S.L.-V.; Dauwerse, J.G.; Baelde, H.J.; Leonhard, W.N.; van de Wal, A.; Ward, C.J.; Verbeek, S.; DeRuiter, M.C.; Breuning, M.H.; de Heer, E.; et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum. Mol. Genet. 2004, 13, 3069–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, A.Y.; Zhang, T.; Michaeel, A.; Blumenfeld, J.; Liu, G.; Zhang, W.; Zhang, Z.; Zhu, Y.; Rennert, L.; Martin, C.; et al. Somatic Mutations in Renal Cyst Epithelium in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 2139–2156. [Google Scholar] [CrossRef]
- Koptides, M.; Hadjimichael, C.; Koupepidou, P.; Pierides, A.; Deltas, C.C. Germinal and somatic mutations in the PKD2 gene of renal cysts in autosomal dominant polycystic kidney disease. Hum. Mol. Genet. 1999, 8, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Conduit, S.E.; Davies, E.M.; Ooms, L.M.; Gurung, R.; McGrath, M.; Hakim, S.; Cottle, D.L.; Smyth, I.; Dyson, J.M.; Mitchell, C.A. AKT signaling promotes DNA damage accumulation and proliferation in polycystic kidney disease. Hum. Mol. Genet. 2020, 29, 31–48. [Google Scholar] [CrossRef]
- Li, M.; Qin, S.; Wang, L.; Zhou, J. Genomic instability in patients with autosomal-dominant polycystic kidney disease. J. Int. Med Res. 2013, 41, 169–175. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Saravanabavan, S.; Chandra, A.N.; Munt, A.; Wong, A.T.; Harris, P.C.; Harris, D.C.; McKenzie, P.; Wang, Y.; Rangan, G.K. Up-Regulation of DNA Damage Response Signaling in Autosomal Dominant Polycystic Kidney Disease. Am. J. Pathol. 2021, 191, 902–920. [Google Scholar] [CrossRef]
- Ta, M.H.; Schwensen, K.G.; Liuwantara, D.; Huso, D.L.; Watnick, T.; Rangan, G.K. Constitutive renal Rel/nuclear factor-κB expression in Lewis polycystic kidney disease rats. World J. Nephrol. 2016, 5, 339–357. [Google Scholar] [CrossRef]
- Zhang, J.Q.J.; Saravanabavan, S.; Munt, A.; Wong, A.T.Y.; Harris, D.C.; Harris, P.C.; Wang, Y.; Rangan, G.K. The role of DNA damage as a therapeutic target in autosomal dominant polycystic kidney disease. Expert Rev. Mol. Med. 2019, 21, e6. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xu, X.; Chen, Y.; Cheung, J.C.; Wang, H.; Jiang, J.; de Val, N.; Fox, T.; Gellert, M.; Yang, W. Structure of an activated DNA-PK and its implications for NHEJ. Mol. Cell 2021, 81, 801–810.e3. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Huang, E.Y.-H.; Huang, S.-C.; Chung, B.-C. DNA-PK/Chk2 induces centrosome amplification during prolonged replication stress. Oncogene 2015, 34, 1263–1269. [Google Scholar] [CrossRef]
- Chen, T.-Y.; Huang, B.-M.; Tang, T.K.; Chao, Y.-Y.; Xiao, X.-Y.; Lee, P.-R.; Yang, L.-Y.; Wang, C.-Y. Genotoxic stress-activated DNA-PK-p53 cascade and autophagy cooperatively induce ciliogenesis to maintain the DNA damage response. Cell Death Differ. 2021, 28, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Gavrilova, O.; Brown, A.L.; Soto, J.A.; Bremner, S.; Kim, J.; Xu, X.; Yang, S.; Um, J.-H.; Koch, L.G.; et al. DNA-PK Promotes the Mitochondrial, Metabolic, and Physical Decline that Occurs During Aging. Cell Metab. 2017, 25, 1135–1146.e7. [Google Scholar] [CrossRef] [Green Version]
- Dionne, L.K.; Shim, K.; Hoshi, M.; Cheng, T.; Wang, J.; Marthiens, V.; Knoten, A.; Basto, R.; Jain, S.; Mahjoub, M.R. Centrosome amplification disrupts renal development and causes cystogenesis. J. Cell Biol. 2018, 217, 2485–2501. [Google Scholar] [CrossRef] [Green Version]
- Padovano, V.; Podrini, C.; Boletta, A.; Caplan, M.J. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat. Rev. Nephrol. 2018, 14, 678–687. [Google Scholar] [CrossRef]
- Nowak, K.L.; Hopp, K. Metabolic Reprogramming in Autosomal Dominant Polycystic Kidney Disease: Evidence and Therapeutic Potential. Clin. J. Am. Soc. Nephrol. 2020, 15, 577–584. [Google Scholar] [CrossRef]
- Battini, L.; Macip, S.; Fedorova, E.; Dikman, S.; Somlo, S.; Montagna, C.; Gusella, G.L. Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum. Mol. Genet. 2008, 17, 2819–2833. [Google Scholar] [CrossRef] [Green Version]
- Booij, T.; Bange, H.; Leonhard, W.; Yan, K.; Fokkelman, M.; Kunnen, S.; Dauwerse, J.G.; Qin, Y.; Van De Water, B.; van Westen, G.; et al. High-Throughput Phenotypic Screening of Kinase Inhibitors to Identify Drug Targets for Polycystic Kidney Disease. SLAS Discov. Adv. Life Sci. R&D 2017, 22, 974–984. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Kim, J.; Schrier, R.W.; Edelstein, C.L. Rapamycin Markedly Slows Disease Progression in a Rat Model of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2005, 16, 46–51. [Google Scholar] [CrossRef] [Green Version]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Wahl, P.R.; Le Hir, M.; Wäckerle-Men, Y.; Wüthrich, R.P.; Serra, A.L. Everolimus Retards Cyst Growth and Preserves Kidney Function in a Rodent Model for Polycystic Kidney Disease. Kidney Blood Press. Res. 2007, 30, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.L.; Poster, D.; Kistler, A.D.; Krauer, F.; Raina, S.; Young, J.; Rentsch, K.M.; Spanaus, K.S.; Senn, O.; Kristanto, P.; et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2010, 363, 820–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walz, G.; Budde, K.; Mannaa, M.; Nürnberger, J.; Wanner, C.; Sommerer, C.; Kunzendorf, U.; Banas, B.; Hörl, W.H.; Obermüller, N.; et al. Everolimus in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2010, 363, 830–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Di Giovanni, V.; He, N.; Wang, K.; Ingram, A.; Rosenblum, N.D.; Pei, Y. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): Computational identification of gene ex-pression pathways and integrated regulatory networks. Hum. Mol. Genet. 2009, 18, 2328–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rödder, S.; Scherer, A.; Raulf, F.; Berthier, C.C.; Hertig, A.; Couzi, L.; Durrbach, A.; Rondeau, E.; Marti, H.-P. Renal Allografts with IF/TA Display Distinct Expression Profiles of Metzincins and Related Genes. Am. J. Transplant. 2009, 9, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Mezard, P.; Berthier, C.C.; Zhang, H.; Hertig, A.; Kaiser, S.; Schumacher, M.; Wieczorek, G.; Bigaud, M.; Kehren, J.; Rondeau, E.; et al. Analysis of independent microarray datasets of renal biopsies identifies a robust transcript signature of acute allograft rejection. Transpl. Int. 2009, 22, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Loghman-Adham, M.; Nauli, S.M.; Soto, C.E.; Kariuki, B.; Zhou, J. Immortalized epithelial cells from human autosomal dominant polycystic kidney cysts. Am. J. Physiol. Renal Physiol. 2003, 285, F397–F412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Mao, J.-H.; Li, X.-Q.; Qian, L.; Zhu, H.; Gu, D.-H.; Pan, X.-D. Over-expression of DNA-PKcs in renal cell carcinoma regulates mTORC2 activation, HIF-2α expression and cell proliferation. Sci. Rep. 2016, 6, 29415. [Google Scholar] [CrossRef] [PubMed]
- Jan, Y.-H.; Heck, D.E.; Laskin, D.L.; Laskin, J.D. Sulfur Mustard Analog Mechlorethamine (Bis(2-chloroethyl)methylamine) Modulates Cell Cycle Progression via the DNA Damage Response in Human Lung Epithelial A549 Cells. Chem. Res. Toxicol. 2019, 32, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Leahy, J.J.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Richardson, C.; Rigoreau, L.; Smith, G.C. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg. Med. Chem. Lett. 2004, 14, 6083–6087. [Google Scholar] [CrossRef]
- Meier-Kriesche, H.-U.; Kaplan, B. Toxicity and efficacy of sirolimus: Relationship to whole-blood concentrations. Clin. Ther. 2000, 22 Suppl. B, B93–B100. [Google Scholar] [CrossRef]
- Novalic, Z.; Van Der Wal, A.M.; Leonhard, W.; Koehl, G.; Breuning, M.H.; Geissler, E.K.; De Heer, E.; Peters, D.J. Dose-Dependent Effects of Sirolimus on mTOR Signaling and Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2012, 23, 842–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choo, S.; Chowbay, B.; Ng, Q.; Thng, C.; Lim, C.; Hartono, S.; Koh, T.; Huynh, H.; Poon, D.; Ang, M.; et al. A Phase 1 dose-finding and pharmacodynamic study of rapamycin in combination with bevacizumab in patients with unresectable hepatocellular carcinoma. Eur. J. Cancer 2013, 49, 999–1008. [Google Scholar] [CrossRef]
- Holditch, S.J.; Brown, C.N.; Atwood, D.J.; Lombardi, A.M.; Nguyen, K.N.; Toll, H.W.; Hopp, K.; Edelstein, C.L. A study of sirolimus and mTOR kinase inhibitor in a hypomorphic Pkd1 mouse model of autosomal dominant polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2019, 317, F187–F196. [Google Scholar] [CrossRef]
- Zhang, J.Q.J.; Saravanabavan, S.; Rangan, G.K. Effect of Reducing Ataxia-Telangiectasia Mutated (ATM) in Experimental Autosomal Dominant Polycystic Kidney Disease. Cells 2021, 10, 532. [Google Scholar] [CrossRef]
- Sunada, S.; Kanai, H.; Lee, Y.; Yasuda, T.; Hirakawa, H.; Liu, C.; Fujimori, A.; Uesaka, M.; Okayasu, R. Nontoxic concentration of DNA—PK inhibitor NU7441 radio-sensitizes lung tumor cells with little effect on double strand break repair. Cancer Sci. 2016, 107, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Timme, C.R.; Rath, B.H.; O’neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and as Orthotopic Xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, H.C.; Iyer, G.V.; Moore, K.; Temkin, S.M.; Gordon, S.; Aghajanian, C.; Grisham, R.N. Activity of M3814, an Oral DNA-PK Inhibitor, In Combination with Topoisomerase II Inhibitors in Ovarian Cancer Models. Sci. Rep. 2019, 9, 18882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [Green Version]
- Willoughby, C.E.; Jiang, Y.; Thomas, H.D.; Willmore, E.; Kyle, S.; Wittner, A.; Phillips, N.; Zhao, Y.; Tudhope, S.J.; Prendergast, L.; et al. Selective DNA-PKcs inhibition extends the therapeutic index of localized radiotherapy and chemotherapy. J. Clin. Investig. 2020, 130, 258–271. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Tissue | Fold Change * | |
|---|---|---|---|
| Average | 95% Confidence Interval | ||
| PRKDC | Normal (n = 13) | 1.00 | (0.80, 1.25) |
| ADPKD (n = 18) | 2.12 a | (1.78, 2.53) | |
| Minimally cystic (n = 5) | 1.41 c | (1.13, 1.76) | |
| Small cysts (<1 mL; n = 5) | 2.04 b | (1.41, 2.95) | |
| Medium cysts (10–25 mL; n = 5) | 2.61 a | (1.17, 3.05) | |
| Large cysts (>50 mL; n =3) | 3.18 a | (1.41, 4.47) | |
| XRCC5 | Normal (n = 13) | 1.00 | (0.84, 1.19) |
| ADPKD (n = 18) | 1.79 b | (1.68, 1.90) | |
| Minimally cystic (n = 5) | 1.72 b | (1.22, 2.10) | |
| Small cysts (<1 mL; n = 5) | 1.83 a | (1.51, 2.19) | |
| Medium cysts (10–25 mL; n = 5) | 1.71 b | (1.51, 1.93) | |
| Large cysts (>50 mL; n = 3) | 1.97 a | (1.75, 2.22) | |
| XRCC6 | Normal (n = 13) | 1.00 | (0.89, 1.12) |
| ADPKD (n = 18) | 1.65 a | (1.53, 1.78) | |
| Minimally cystic (n = 5) | 1.45 c | (1.15, 1.82) | |
| Small cysts (<1 mL; n = 5) | 1.63 a | (1.48, 1.80) | |
| Medium cysts (10–25 mL; n = 5) | 1.81 a | (1.53, 2.15) | |
| Large cysts (>50 mL; n = 3) | 1.77 a | (1.58, 1.98) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandra, A.N.; Saravanabavan, S.; Rangan, G.K. Role of DNA-Dependent Protein Kinase in Mediating Cyst Growth in Autosomal Dominant Polycystic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 10512. https://doi.org/10.3390/ijms221910512
Chandra AN, Saravanabavan S, Rangan GK. Role of DNA-Dependent Protein Kinase in Mediating Cyst Growth in Autosomal Dominant Polycystic Kidney Disease. International Journal of Molecular Sciences. 2021; 22(19):10512. https://doi.org/10.3390/ijms221910512
Chicago/Turabian StyleChandra, Ashley N., Sayanthooran Saravanabavan, and Gopala K. Rangan. 2021. "Role of DNA-Dependent Protein Kinase in Mediating Cyst Growth in Autosomal Dominant Polycystic Kidney Disease" International Journal of Molecular Sciences 22, no. 19: 10512. https://doi.org/10.3390/ijms221910512