
N6-Isopentenyladenosine Hinders the Vasculogenic Mimicry in Human Glioblastoma Cells through Src-120 Catenin Pathway Modulation and RhoA Activity Inhibition

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
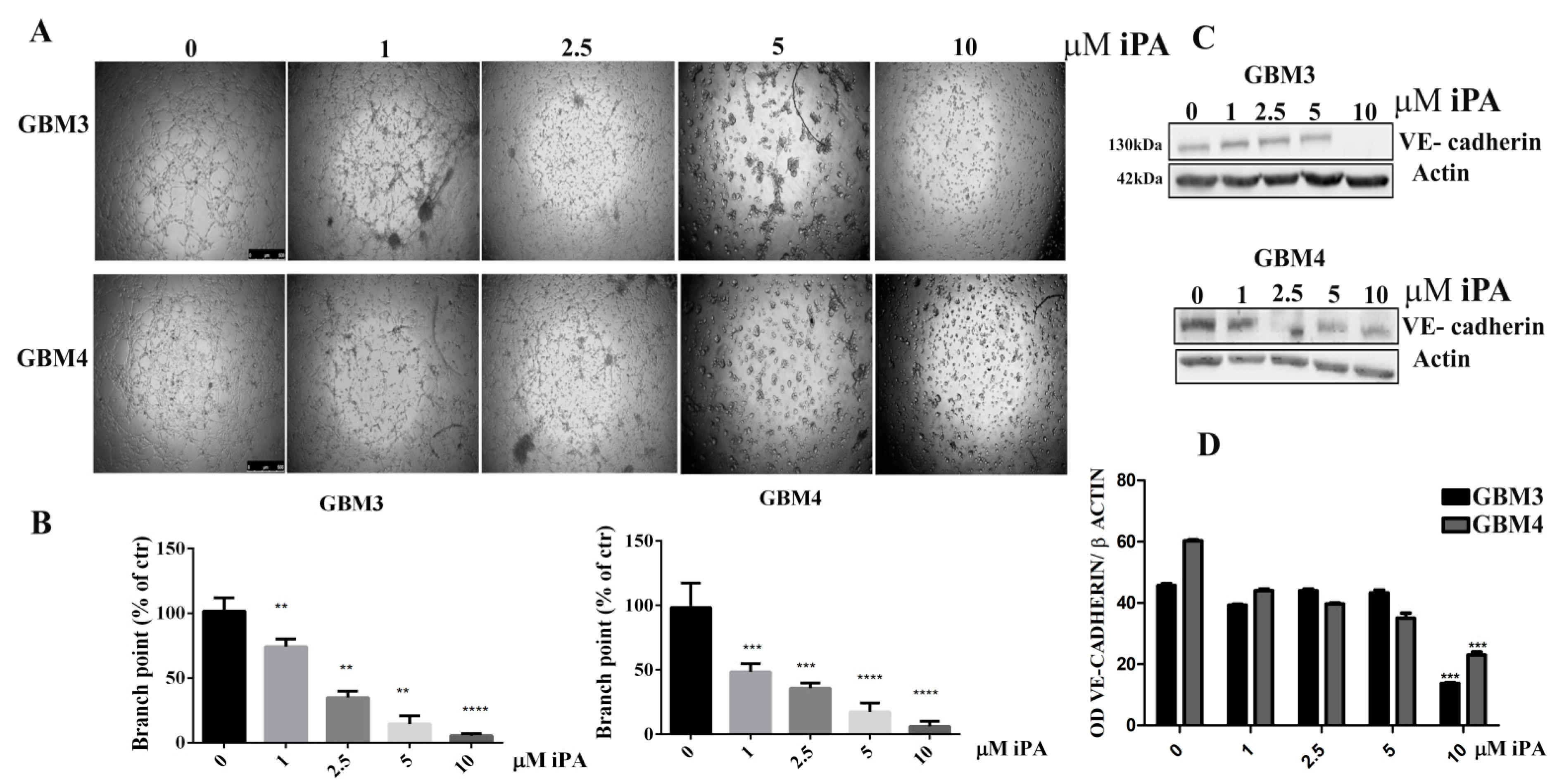
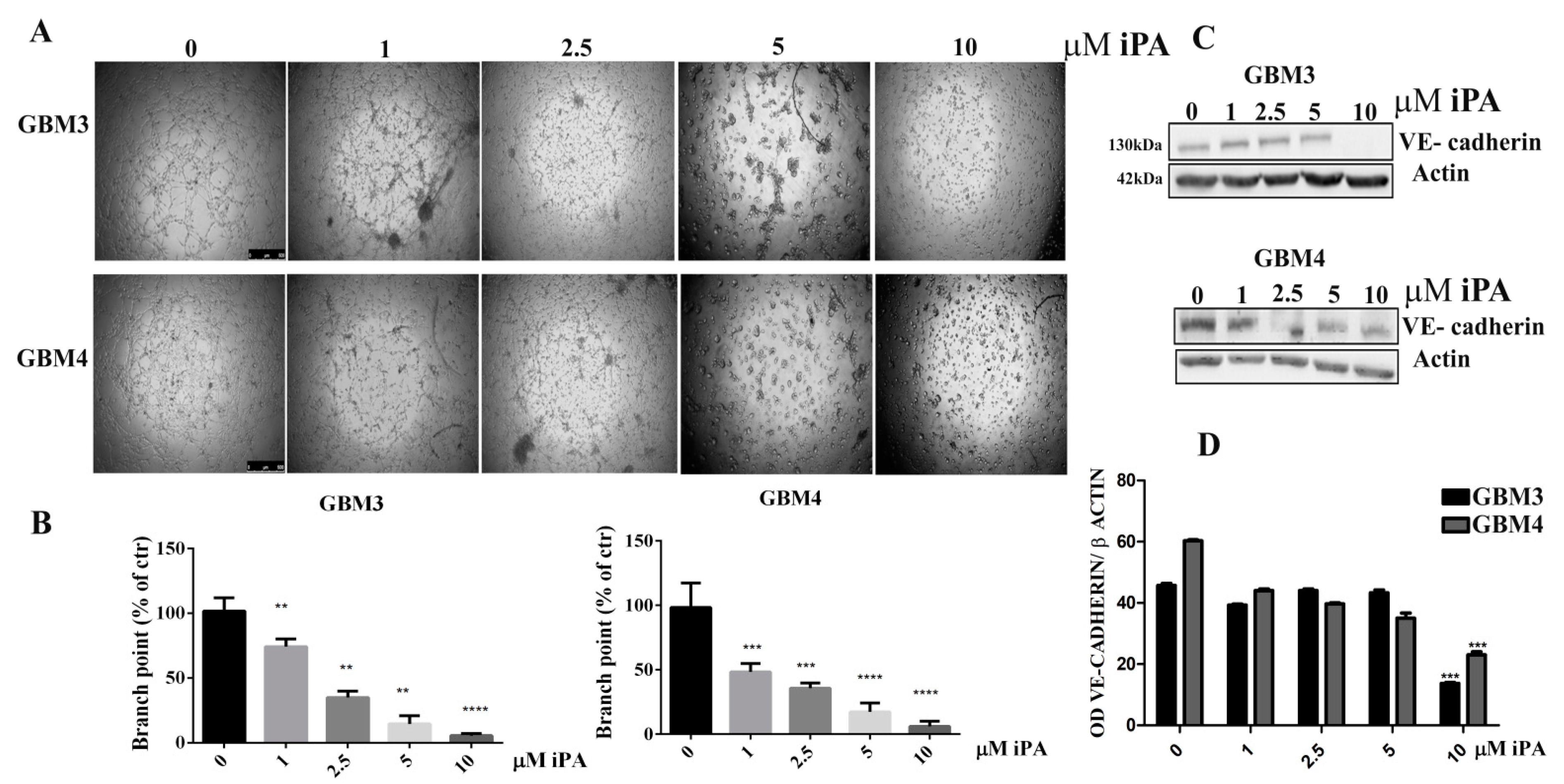
2.1. N6-Isopentenyladenosine Inhibits the Tube Formation in GBM Cells
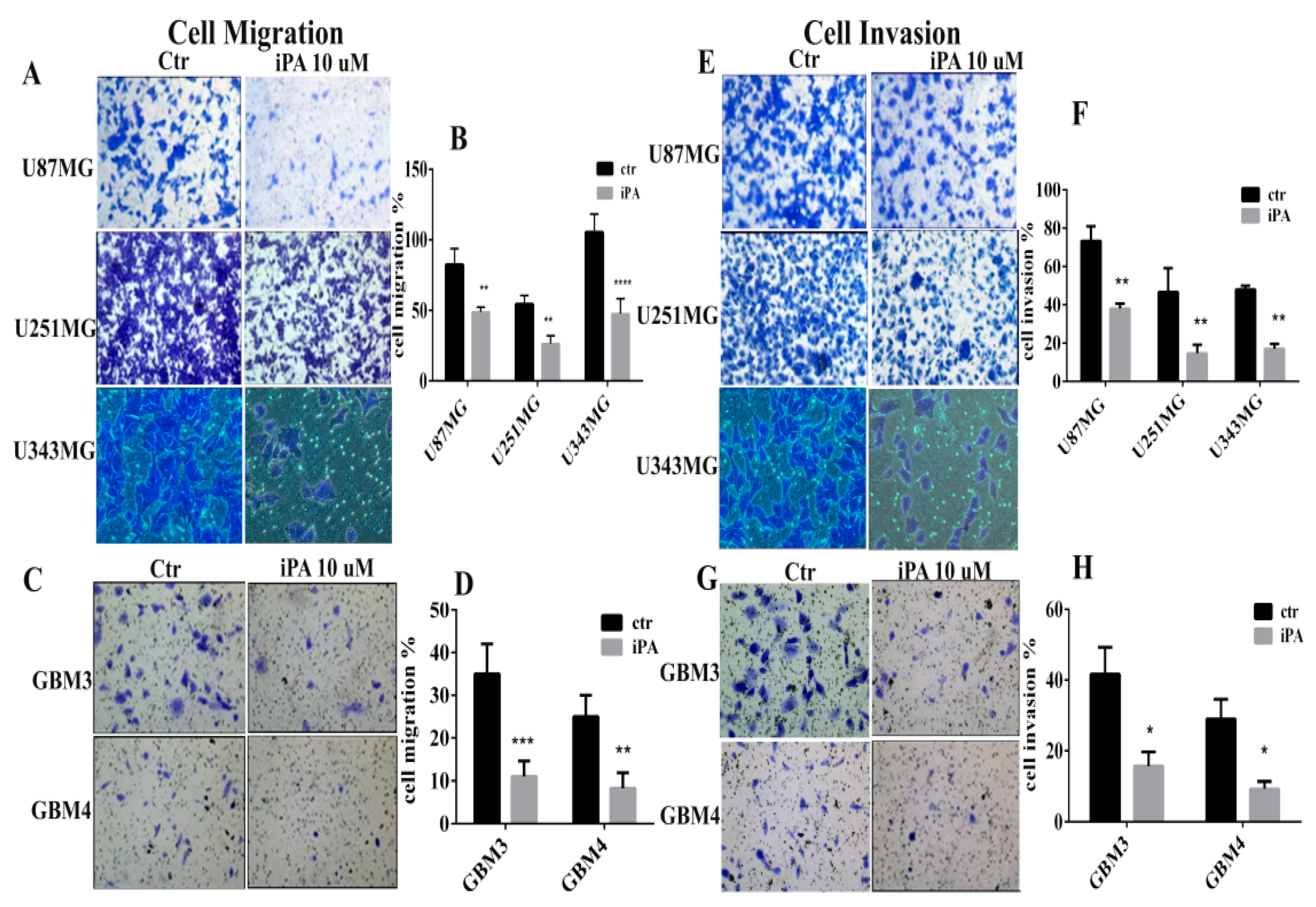
2.2. Inhibitory Effect of iPA on Cell Motility In Vitro
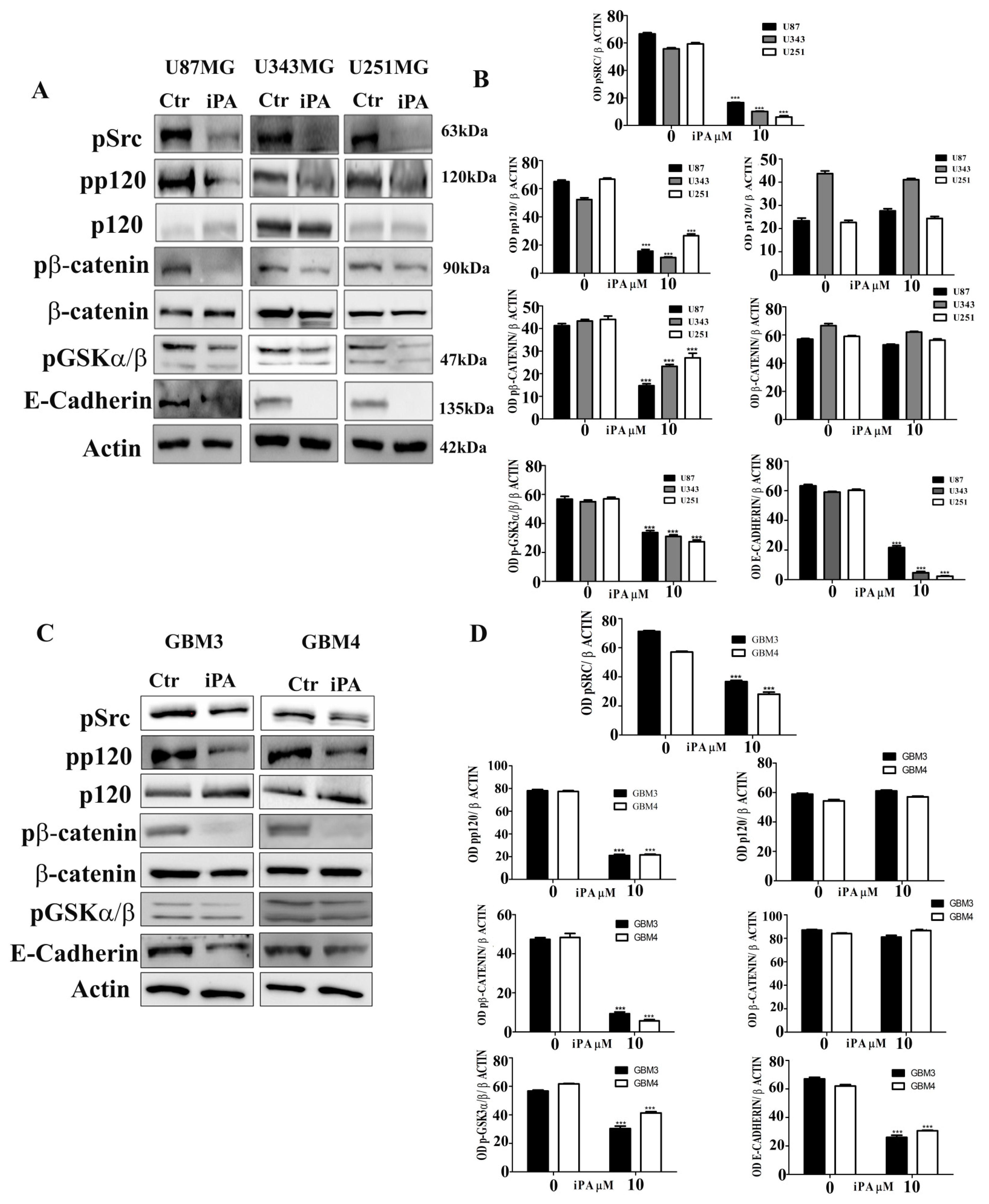
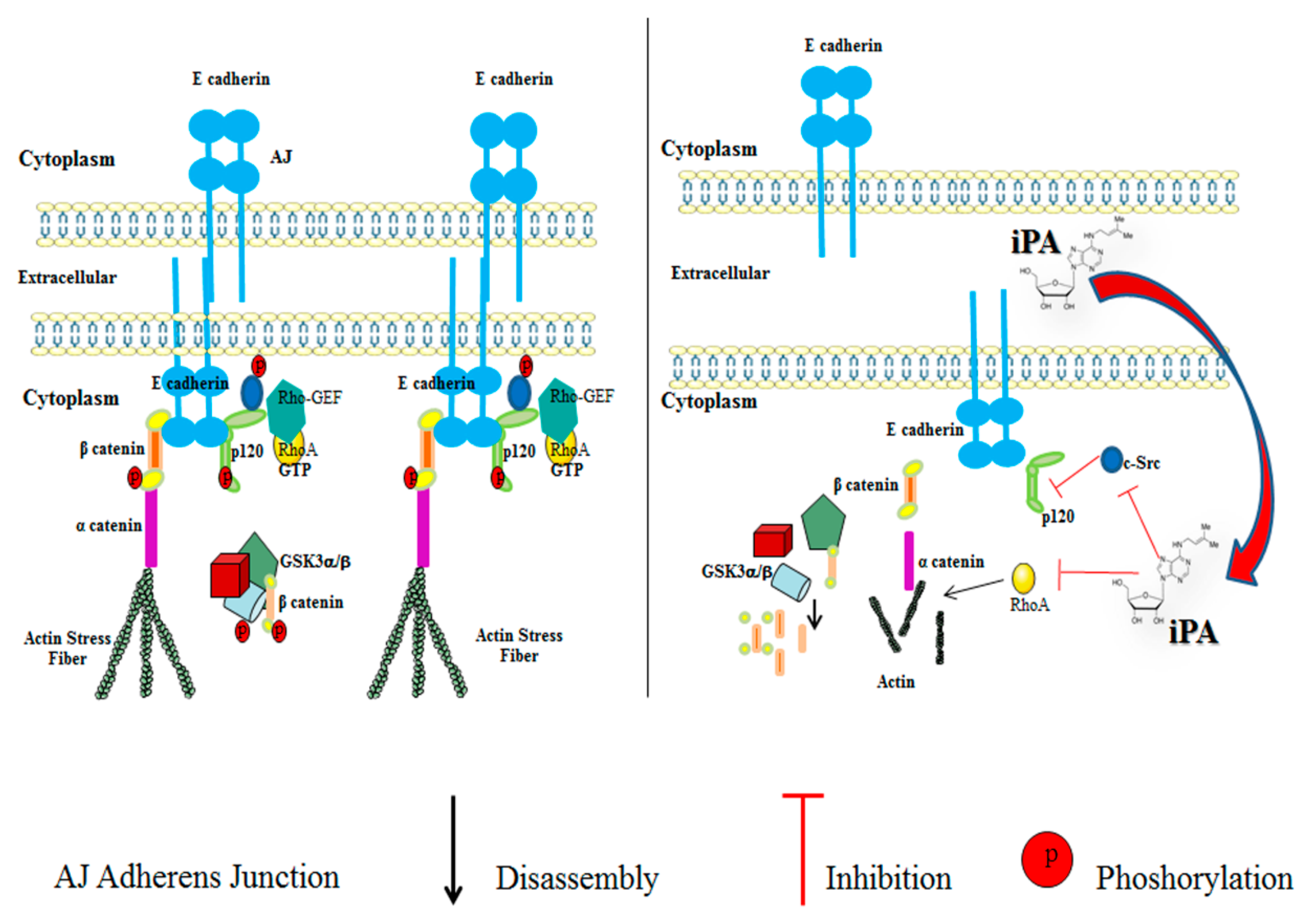
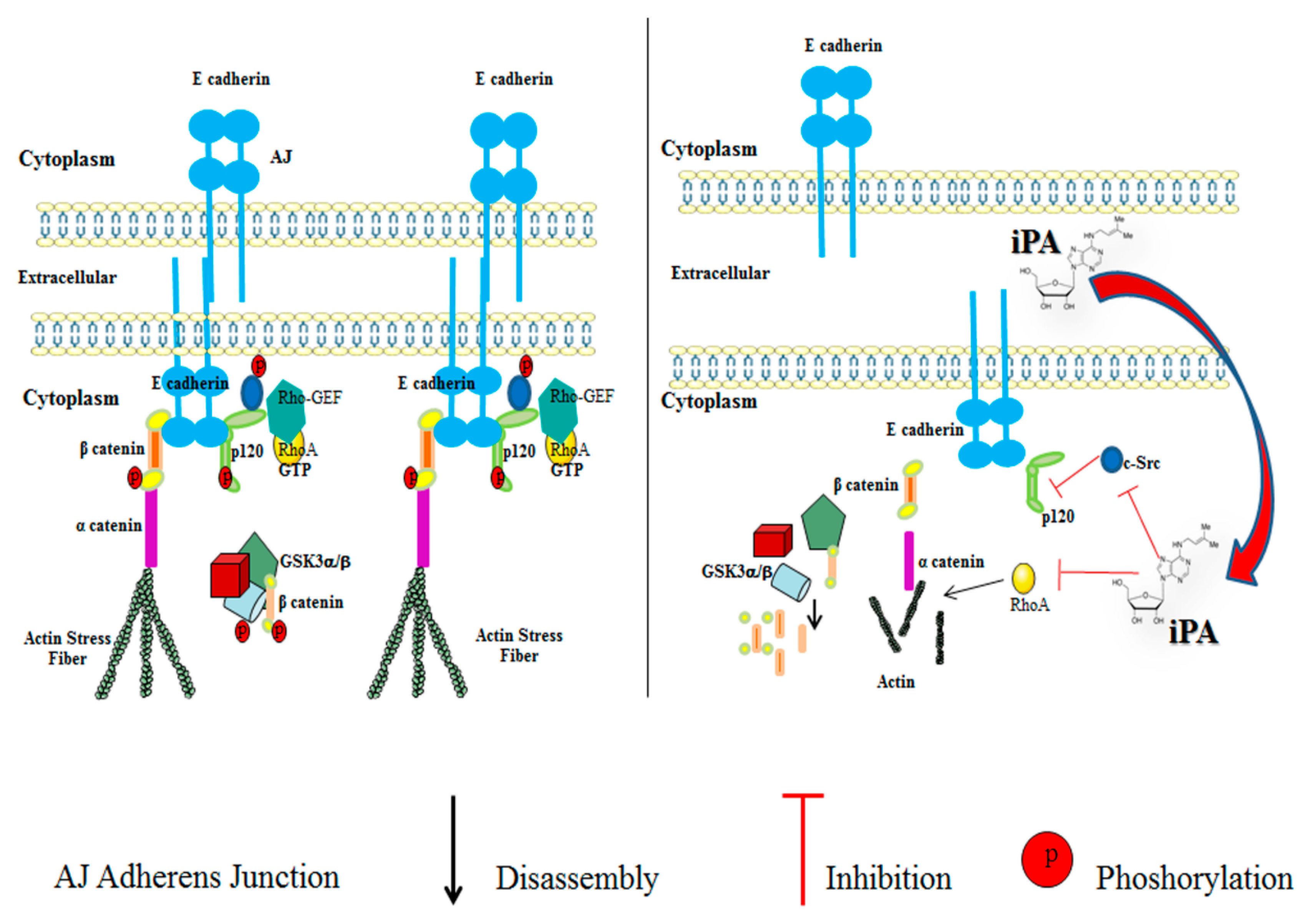
2.3. Effect of iPA on Src-120 Catenin Pathway
2.4. Effect of iPA on RhoA GTPase Activity
3. Discussion
4. Materials and Methods
4.1. Drugs and Reagents
4.2. Cell Cultures
4.3. Primary Glioblastoma Characterization
4.4. IDH1 and IDH2 Mutation Status
4.5. MGMT Methylation Assessment
4.6. Reagents and Antibodies
4.7. MTT Vitality Assay
4.8. Western Blot Analysis
4.9. Cell Migration and Invasion Assay
4.10. In Vitro Tube Formation Assay
4.11. Subcellular Fractionation
4.12. Immunofluorescence Staining
4.13. Active Rho Pulldown Assay
4.14. Immunoprecipitation
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3D | Three-dimensional |
| AJ | Adherens Junction |
| AMPK | 5’ adenosine monophosphate-activated protein kinase |
| ANOVA | Analysis of variance |
| BSA | Bovine serum albumin |
| Cdc42 | Cell division control protein 42 |
| CSCs | Cancer stem cells |
| c-Src | Proto-oncogene tyrosine-protein kinase Src |
| DAPI | 6-diamidino-2-phenylindole |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| DMSO | Dimethyl sulfoxide |
| E-Cadherin | Epithelial cadherin |
| ECM | Extracellular Matrix |
| EGFR | Epidermal growth factor receptor |
| FBS | Fetal bovine serum |
| FFPE | Formalin-fixed, paraffin-embedded |
| FITC | Fluorescein isothiocyanate |
| FPP | Farnesylpyrophosphate |
| GBM CSC | Glioma stem cell lines |
| GBM | Glioblastoma |
| GGPP | Geranylgeranylpyrophosphate |
| GSK | Glycogen synthase kinase |
| GST | Glutathione S-transferase |
| HCC | Human hepatocellular carcinoma |
| HRP | Horseradish peroxidase |
| HUVEC | Human umbilical vein endothelial cells |
| IDH1/2 | Isocitrate dehydrogenase (NADP(+)) 1⁄2 |
| iPA | N6-isopentenyladenosine |
| MGMT | O6-methylguanine DNA methyltransferase |
| MSP | Methylation-specific PCR |
| MTT | 3-(4,5-dimethylthiazol-2yl)-2,5-diphenylterazolium bromide |
| PBS | Phosphate-buffered saline |
| PCR | Polymerase chain reaction |
| RhoA | Ras homolog family member A |
| Rhotekin-RBD | Rho binding domain (RBD) of the human Rhotekin |
| RIPA Buffer | Radioimmunoprecipitation assay buffer |
| ROCK | Rho-associated protein kinase |
| SD | Standard deviation |
| SDS-PAGE | Sodium dodecyl sulphate–polyacrylamide gel electrophoresis |
| SF buffer | Subcellular fractionation buffer |
| TRITC | Tetramethylrhodamine |
| VE-Cadherin | Vascular endothelial cadherin |
| VM | Vasculogenic mimicry |
References
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E., 2nd; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J.; et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J. Clin. Oncol. 2007, 25, 4722–4729. [Google Scholar] [CrossRef] [Green Version]
- Kreisl, T.N.; Lassman, A.B.; Mischel, P.S.; Rosen, N.; Scher, H.I.; Teruya-Feldstein, J.; Shaffer, D.; Lis, E.; Abrey, L.E. A pilot study of everolimus and gefitinib in the treatment of recurrent glioblastoma (GBM). J. Neurooncol. 2009, 92, 99–105. [Google Scholar] [CrossRef]
- Luo, Q.; Wang, J.; Zhao, W.; Peng, Z.; Liu, X.; Li, B.; Zhang, H.; Shan, B.; Zhang, C.; Duan, C. Vasculogenic mimicry in carcinogenesis and clinical applications. J. Hematol. Oncol. 2020, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Soda, Y.; Myskiw, C.; Rommel, A.; Verma, I.M. Mechanisms of neovascularization and resistance to anti-angiogenic therapies in glioblastoma multiforme. J. Mol. Med. 2013, 91, 439–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, T.; Wu, J.; Hu, Y.; Zhang, M.; He, J. Long non-coding RNA HULC stimulates the epithelial-mesenchymal transition process and vasculogenic mimicry in human glioblastoma. Cancer Med. 2021, 10, 5270–5282. [Google Scholar] [CrossRef] [PubMed]
- Treps, L.; Faure, S.; Clere, N. Vasculogenic mimicry, a complex and devious process favoring tumorigenesis—Interest in making it a therapeutic target. Pharmacol. Ther. 2021, 223, 107805. [Google Scholar] [CrossRef]
- Pokutta, S.; Weis, W.I. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu. Rev. Cell Dev. Biol. 2007, 23, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Spiering, D.; Hodgson, L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh. Migr. 2011, 5, 170–180. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.G.; Zhou, H.M.; Zhang, X.; Mu, W.; Hu, J.N.; Liu, G.L.; Li, Q. Hypoxic induction of vasculogenic mimicry in hepatocellular carcinoma: Role of HIF-1 α, RhoA/ROCK and Rac1/PAK signaling. BMC Cancer 2020, 20, 32. [Google Scholar] [CrossRef]
- Navarra, G.; Pagano, C.; Pacelli, R.; Crescenzi, E.; Longobardi, E.; Gazzerro, P.; Fiore, D.; Pastorino, O.; Pentimalli, F.; Laezza, C.; et al. N6-Isopentenyladenosine Enhances the Radiosensitivity of Glioblastoma Cells by Inhibiting the Homologous Recombination Repair Protein RAD51 Expression. Front. Oncol. 2020, 9, 1498. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, R.; Ciaglia, E.; Amodio, G.; Picardi, P.; Proto, M.C.; Gazzerro, P.; Laezza, C.; Remondelli, P.; Bifulco, M.; Pisanti, S. N6-isopentenyladenosine dual targeting of AMPK and Rab7 prenylation inhibits melanoma growth through the impairment of autophagic flux. Cell Death Differ. 2018, 25, 353–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laezza, C.; Malfitano, A.M.; Di Matola, T.; Ricchi, P.; Bifulco, M. Involvement of Akt/NF-κB pathway in N6-isopentenyladenosine-induced apoptosis in human breast cancer cells. Mol. Carcinog. 2010, 49, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Ciaglia, E.; Laezza, C.; Abate, M.; Pisanti, S.; Ranieri, R.; D’alessandro, A.; Picardi, P.; Gazzerro, P.; Bifulco, M. Recognition by natural killer cells of N6-isopentenyladenosine-treated human glioma cell lines. Int. J. Cancer 2018, 142, 176–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciaglia, E.; Abate, M.; Laezza, C.; Pisanti, S.; Vitale, M.; Seneca, V.; Torelli, G.; Franceschelli, S.; Catapano, G.; Gazzerro, P.; et al. Antiglioma effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, through the downregulation of epidermal growth factor receptor. Int. J. Cancer 2017, 140, 959–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisanti, S.; Picardi, P.; Ciaglia, E.; Margarucci, L.; Ronca, R.; Giacomini, A.; Malfitano, A.M.; Casapullo, A.; Laezza, C.; Gazzerro, P.; et al. Antiangiogenic effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, mediated by AMPK activation. FASEB J. 2014, 28, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, O.; Gentile, M.T.; Mancini, A.; Del Gaudio, N.; Di Costanzo, A.; Bajetto, A.; Franco, P.; Altucci, L.; Florio, T.; Stoppelli, M.P.; et al. Histone Deacetylase Inhibitors Impair Vasculogenic Mimicry from Glioblastoma Cells. Cancers 2019, 11, 747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, J.M.; Liu, J.; Guo, G.; Mao, X.G.; Li, C.X. Glioblastoma vasculogenic mimicry: Signaling pathways progression and potential anti-angiogenesis targets. Biomark. Res. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, J.; Zhou, H.; Fan, G.; Li, Q. Molecular Mechanisms and Anticancer Therapeutic Strategies in Vasculogenic Mimicry. J. Cancer 2019, 10, 6327–6340. [Google Scholar] [CrossRef]
- Barreno, L.; Cáceres, S.; Alonso-Diez, Á.; Vicente-Montaña, A.; García, M.L.; Clemente, M.; Illera, J.C.; Peña, L. Vasculogenic mimicry-associated ultrastructural findings in human and canine inflammatory breast cancer cell lines. BMC Cancer 2019, 19, 750. [Google Scholar] [CrossRef] [Green Version]
- Gloushankova, N.A.; Rubtsova, S.N.; Zhitnyak, I.Y. Cadherin-mediated cell-cell interactions in normal and cancer cells. Tissue Barriers 2017, 5, e1356900. [Google Scholar] [CrossRef] [Green Version]
- Castaño, J.; Solanas, G.; Casagolda, D.; Raurell, I.; Villagrasa, P.; Bustelo, X.R.; García de Herreros, A.; Duñach, M. Specific phosphorylation of p120-catenin regulatory domain differently modulates its binding to RhoA. Mol. Cell. Biol. 2007, 27, 1745–1757. [Google Scholar] [CrossRef] [Green Version]
- Mariner, D.J.; Anastasiadis, P.; Keilhack, H.; Böhmer, F.D.; Wang, J.; Reynolds, A.B. Identification of Src phosphorylation sites in the catenin p120ctn. J. Biol. Chem. 2001, 276, 28006–28013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formisano, L.; D’Amato, V.; Servetto, A.; Brillante, S.; Raimondo, L.; Di Mauro, C.; Marciano, R.; Orsini, R.C.; Cosconati, S.; Randazzo, A.; et al. Src inhibitors act through different mechanisms in Non-Small Cell Lung Cancer models depending on EGFR and RAS mutational status. Oncotarget 2015, 6, 26090–26103. [Google Scholar] [CrossRef] [PubMed]
- Haga, R.B.; Ridley, A.J. Rho GTPases: Regulation and roles in cancer cell biology. Small GTPases 2016, 7, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizaki, T.; Uehata, M.; Tamechika, I.; Keel, J.; Nonomura, K.; Maekawa, M.; Narumiya, S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol. Pharmacol. 2000, 57, 976–983. [Google Scholar] [PubMed]
- Just, I.; Wilm, M.; Selzer, J.; Rex, G.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J. Biol. Chem. 1995, 270, 13932–13936. [Google Scholar] [CrossRef] [Green Version]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santarelli, J.G.; Udani, V.; Yung, Y.C.; Cheshier, S.; Wagers, A.; Brekken, R.A.; Weissman, I.; Tse, V. Incorporation of bone marrow-derived Flk-1-expressing CD34+ cells in the endothelium of tumor vessels in the mouse brain. Neurosurgery 2006, 59, 374–382, discussion 374–382. [Google Scholar] [CrossRef]
- Kurz, H.; Burri, P.H.; Djonov, V.G. Angiogenesis and vascular remodeling by intussusception: From form to function. News Physiol. Sci. 2003, 18, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Bellido, D.; Serrano-Saenz, S.; Fernández-Cortés, M.; Oliver, F.J. Vasculogenic mimicry signaling revisited: Focus on non-vascular VE-cadherin. Mol. Cancer 2017, 16, 65. [Google Scholar] [CrossRef] [Green Version]
- Etienne-Manneville, S. Adherens junctions during cell migration. Subcell Biochem. 2012, 60, 225–249. [Google Scholar]
- Kourtidis, A.; Ngok, S.P.; Anastasiadis, P.Z. p120 catenin: An essential regulator of cadherin stability, adhesion-induced signaling, and cancer progression. Prog. Mol. Biol. Transl. Sci. 2013, 116, 409–432. [Google Scholar]
- Wadhawan, A.; Smith, C.; Nicholson, R.I.; Barrett-Lee, P.; Hiscox, S. Src-mediated regulation of homotypic cell adhesion: Implications for cancer progression and opportunities for therapeutic intervention. Cancer Treat Rev. 2011, 37, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Huveldt, D.; Lewis-Tuffin, L.J.; Carlson, B.L.; Schroeder, M.A.; Rodriguez, F.; Giannini, C.; Galanis, E.; Sarkaria, J.N.; Anastasiadis, P.Z. Targeting Src family kinases inhibits bevacizumab-induced glioma cell invasion. PLoS ONE 2013, 8, e56505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, M.T.; Mo, R.; Flozak, A.S.; Peled, O.N.; Gottardi, C.J. Beta-catenin phosphorylated at serine 45 is spatially uncoupled from beta-catenin phosphorylated in the GSK3 domain: Implications for signaling. PLoS ONE 2010, 5, e10184. [Google Scholar] [CrossRef]
- Noren, N.K.; Liu, B.P.; Burridge, K.; Kreft, B. p120 catenin regulates the actin cytoskeleton via Rho family GTPases. J. Cell Biol. 2000, 150, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.K.; Le, A.; Vijayan, K.V.; Thiagarajan, P. Dasatinib inhibits actin fiber reorganization and promotes endothelial cell permeability through RhoA-ROCK pathway. Cancer Med. 2017, 6, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laezza, C.; Notarnicola, M.; Caruso, M.G.; Messa, C.; Macchia, M.; Bertini, S.; Minutolo, F.; Portella, G.; Fiorentino, L.; Stingo, S.; et al. N6-isopentenyladenosine arrests tumor cell proliferation by inhibiting farnesyl diphosphate synthase and protein prenylation. FASEB J. 2006, 20, 412–418. [Google Scholar] [CrossRef]
- Leve, F.; Marcondes, T.G.; Bastos, L.G.; Rabello, S.V.; Tanaka, M.N.; Morgado-Díaz, J.A. Lysophosphatidic acid induces a migratory phenotype through a crosstalk between RhoA-Rock and Src-FAK signalling in colon cancer cells. Eur. J. Pharmacol. 2011, 671, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiadis, P.Z.; Reynolds, A.B. Regulation of Rho GTPases by p120-catenin. Curr. Opin. Cell Biol. 2001, 13, 604–610. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GBM | Patient | Type | MGMT Methylation Status | IDH1/IDH2 Status | Co-del 1p-19q | Molecular Characterization | EGFR Amplification | Epigenetic Sub-Class | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | Gender | Patient | GBM Line | Patient | GBM Line | ||||||
| GBM3 | 54 | M | Primary | Unmethylated | Unmethylated | IDH WT | IDH WT | absent | GBM IDH WT | yes | RTKII |
| GBM4 | 64 | M | Primary | Unmethylated | Methylated | IDH WT | IDH WT | absent | GBM IDH WT | no | Mesenchymal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, C.; Navarra, G.; Pastorino, O.; Avilia, G.; Coppola, L.; Della Monica, R.; Chiariotti, L.; Florio, T.; Corsaro, A.; Torelli, G.; et al. N6-Isopentenyladenosine Hinders the Vasculogenic Mimicry in Human Glioblastoma Cells through Src-120 Catenin Pathway Modulation and RhoA Activity Inhibition. Int. J. Mol. Sci. 2021, 22, 10530. https://doi.org/10.3390/ijms221910530
Pagano C, Navarra G, Pastorino O, Avilia G, Coppola L, Della Monica R, Chiariotti L, Florio T, Corsaro A, Torelli G, et al. N6-Isopentenyladenosine Hinders the Vasculogenic Mimicry in Human Glioblastoma Cells through Src-120 Catenin Pathway Modulation and RhoA Activity Inhibition. International Journal of Molecular Sciences. 2021; 22(19):10530. https://doi.org/10.3390/ijms221910530
Chicago/Turabian StylePagano, Cristina, Giovanna Navarra, Olga Pastorino, Giorgio Avilia, Laura Coppola, Rosa Della Monica, Lorenzo Chiariotti, Tullio Florio, Alessandro Corsaro, Giovanni Torelli, and et al. 2021. "N6-Isopentenyladenosine Hinders the Vasculogenic Mimicry in Human Glioblastoma Cells through Src-120 Catenin Pathway Modulation and RhoA Activity Inhibition" International Journal of Molecular Sciences 22, no. 19: 10530. https://doi.org/10.3390/ijms221910530