In Silico Identification of the Complex Interplay between Regulatory SNPs, Transcription Factors, and Their Related Genes in Brassica napus L. Using Multi-Omics Data

 , , , , ,
, , , , ,  , and
, and
Abstract
:
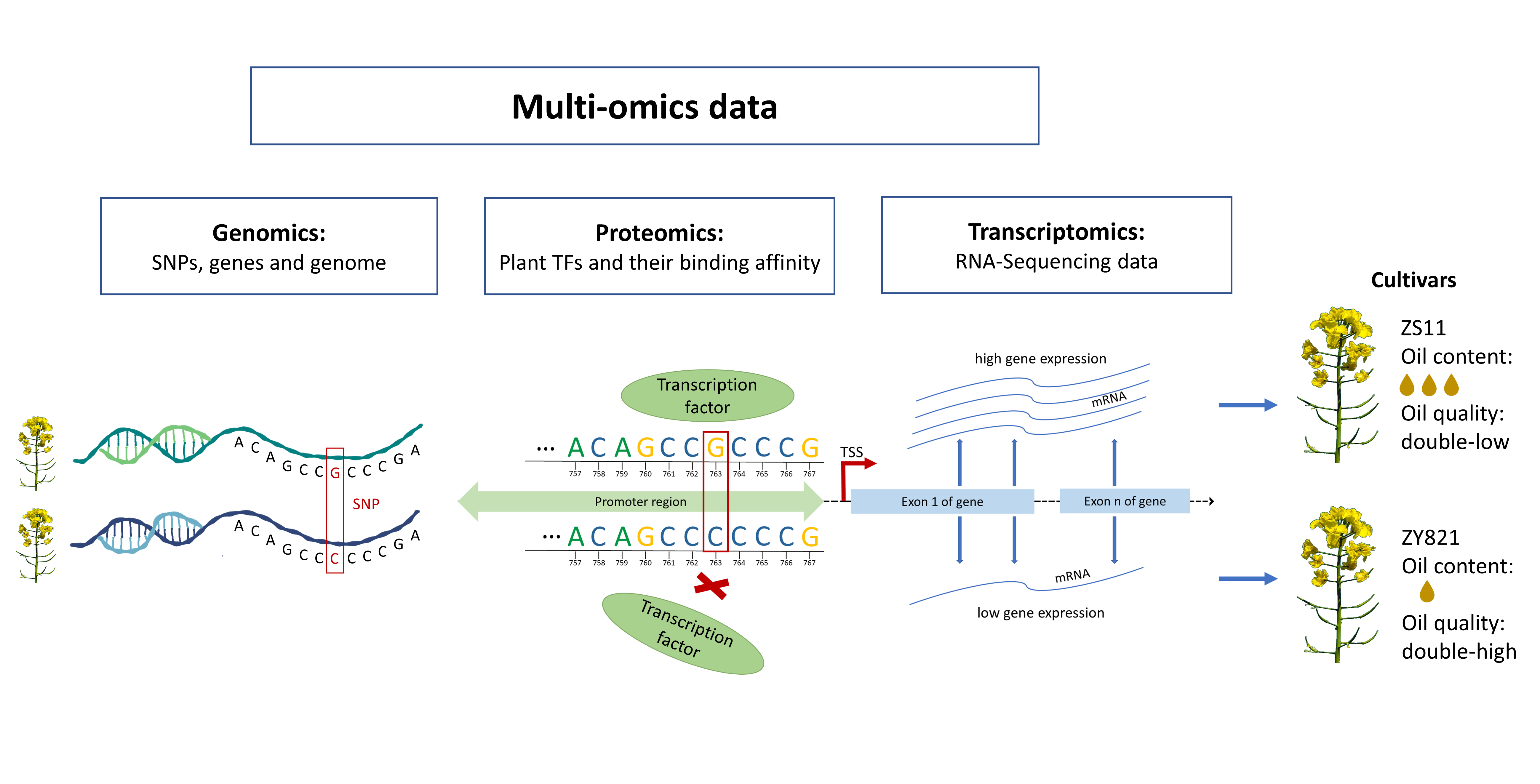
1. Introduction
2. Results and Discussion
2.1. Differentially Expressed Genes
2.2. Transcription Factor Binding Site Enrichment Analysis
2.3. Analysis of Regulatory SNPs
2.4. Analysis of Important Regulatory SNPs
2.5. DEGs Harboring Important rSNPs in the Promoter Region
3. Materials and Methods
3.1. B. napus Data Set and Data Preparation
3.1.1. Genotype Data
3.1.2. Transcriptome Data
3.2. Transcription Factor Binding Site Enrichment Analysis in Promoter Sequences
3.3. Identification of Regulatory SNPs and Their Importance
3.4. Association Analysis Using Random Forests
| Algorithm 1 Boruta Algorithm |
Input:: Genotype (rSNPs) data Input:: Labels (cultivars) Output:: A ranked list of rSNPs based on their importance score Method:
|
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Edwards, S.L.; Beesley, J.; French, J.D.; Dunning, A.M. Beyond GWASs: Illuminating the dark road from association to function. Am. J. Hum. Genet. 2013, 93, 779–797. [Google Scholar] [CrossRef]
- Rojano, E.; Seoane, P.; Ranea, J.A.G.; Perkins, J.R. Regulatory variants: From detection to predicting impact. Briefings Bioinform. 2019, 20, 1639–1654. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, G.; Bailey, J.; Haviv, I.; Kowalczyk, A. is-rSNP: A novel technique for in silico regulatory SNP detection. Bioinformatics 2010, 26, i524–i530. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Weng, J.; Liu, C.; Song, X.; Miao, H.; Hao, Z.; Xie, C.; Li, M.; Zhang, D.; Bai, L.; et al. Identification of promoter motifs regulating ZmeIF4E expression level involved in maize rough dwarf disease resistance in maize (Zea Mays L.). Mol. Genet. Genom. 2013, 288, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Konishi, S.; Izawa, T.; Lin, S.Y.; Ebana, K.; Fukuta, Y.; Sasaki, T.; Yano, M. An SNP caused loss of seed shattering during rice domestication. Science 2006, 312, 1392–1396. [Google Scholar] [CrossRef]
- Jaiswal, V.; Gahlaut, V.; Mathur, S.; Agarwal, P.; Khandelwal, M.K.; Khurana, J.P.; Tyagi, A.K.; Balyan, H.S.; Gupta, P.K. Identification of novel SNP in promoter sequence of TaGW2-6A associated with grain weight and other agronomic traits in wheat (Triticum aestivum L.). PLoS ONE 2015, 10, e0129400. [Google Scholar] [CrossRef]
- Heinrich, F.; Wutke, M.; Das, P.P.; Kamp, M.; Gültas, M.; Link, W.; Schmitt, A.O. Identification of Regulatory SNPs Associated with Vicine and Convicine Content of Vicia faba Based on Genotyping by Sequencing Data Using Deep Learning. Genes 2020, 11, 614. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Zhao, J.; Afek, A.; Mielko, Z.; Gordân, R. QBiC-Pred: Quantitative predictions of transcription factor binding changes due to sequence variants. Nucleic Acids Res. 2019, 47, W127–W135. [Google Scholar] [CrossRef]
- Amlie-Wolf, A.; Tang, M.; Mlynarski, E.E.; Kuksa, P.P.; Valladares, O.; Katanic, Z.; Tsuang, D.; Brown, C.D.; Schellenberg, G.D.; Wang, L.S. INFERNO: Inferring the molecular mechanisms of noncoding genetic variants. Nucleic Acids Res. 2018, 46, 8740–8753. [Google Scholar] [CrossRef]
- Coetzee, S.G.; Coetzee, G.A.; Hazelett, D.J. motifbreakR: An R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 2015, 31, 3847–3849. [Google Scholar] [CrossRef]
- Medina-Rivera, A.; Defrance, M.; Sand, O.; Herrmann, C.; Castro-Mondragon, J.A.; Delerce, J.; Jaeger, S.; Blanchet, C.; Vincens, P.; Caron, C.; et al. RSAT 2015: Regulatory sequence analysis tools. Nucleic Acids Res. 2015, 43, W50–W56. [Google Scholar] [CrossRef] [PubMed]
- Zuo, C.; Shin, S.; Keleş, S. atSNP: Transcription factor binding affinity testing for regulatory SNP detection. Bioinformatics 2015, 31, 3353–3355. [Google Scholar] [CrossRef] [PubMed]
- Lohani, N.; Jain, D.; Singh, M.B.; Bhalla, P.L. Engineering Multiple Abiotic Stress Tolerance in Canola, Brassica napus. Front. Plant Sci. 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Friedt, W.; Snowdon, R. Oilseed Rape. In Oil Crops; Springer: New York, NY, USA, 2009; pp. 91–126. [Google Scholar] [CrossRef]
- Allender, C.J.; King, G.J. Origins of the amphiploid species Brassica napus L. investigated by chloroplast and nuclear molecular markers. BMC Plant Biol. 2010, 10, 54. [Google Scholar] [CrossRef]
- Becker, H. Pflanzenzüchtung; UTB GmbH: Stuttgart, Germany, 2019. [Google Scholar]
- Hatzig, S.V.; Nuppenau, J.N.; Snowdon, R.J.; Schießl, S.V. Drought stress has transgenerational effects on seeds and seedlings in winter oilseed rape (Brassica napus L.). BMC Plant Biol. 2018, 18, 297. [Google Scholar] [CrossRef]
- Gupta, M.; Bhaskar, P.B.; Sriram, S.; Wang, P.H. Integration of omics approaches to understand oil/protein content during seed development in oilseed crops. Plant Cell Rep. 2017, 36, 637–652. [Google Scholar] [CrossRef]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Agrawal, G.K.; Rakwal, R. Seed Development: OMICS Technologies Toward Improvement of Seed Quality and Crop Yield; Springer: Dordrecht, The Netherlands, 2012. [Google Scholar]
- Ramzan, F.; Gültas, M.; Bertram, H.; Cavero, D.; Schmitt, A.O. Combining Random Forests and a Signal Detection Method Leads to the Robust Detection of Genotype-Phenotype Associations. Genes 2020, 11, 892. [Google Scholar] [CrossRef]
- Ramzan, F.; Klees, S.; Schmitt, A.O.; Cavero, D.; Gültas, M. Identification of Age-Specific and Common Key Regulatory Mechanisms Governing Eggshell Strength in Chicken Using Random Forests. Genes 2020, 11, 464. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, G.; Chen, S. Structure and regulatory function of plant transcription factors. Chin. Sci. Bull. 2001, 46, 271–278. [Google Scholar] [CrossRef]
- Zhang, C.; Hou, Y.; Hao, Q.; Chen, H.; Chen, L.; Yuan, S.; Shan, Z.; Zhang, X.; Yang, Z.; Qiu, D.; et al. Genome-wide survey of the soybean GATA transcription factor gene family and expression analysis under low nitrogen stress. PLoS ONE 2015, 10, e0125174. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.C.; Muro-Pastor, M.I.; Florencio, F.J. The GATA family of transcription factors in Arabidopsis and rice. Plant Physiol. 2004, 134, 1718–1732. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Hu, S.; Yu, Q.; Wang, C.; Yang, Y.; Sun, H.; Yang, Y.; Sun, X. Genome-Wide Identification and Characterization of BrrTCP Transcription Factors in Brassica rapa ssp. rapa. Front. Plant Sci. 2017, 8, 1588. [Google Scholar] [CrossRef]
- Martín-Trillo, M.; Cubas, P. TCP genes: A family snapshot ten years later. Trends Plant Sci. 2010, 15, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Jang, I.C.; Henriques, R.; Seo, H.S.; Nagatani, A.; Chua, N.H. Arabidopsis phytochrome interacting factor proteins promote phytochrome B polyubiquitination by COP1 E3 ligase in the nucleus. Plant Cell 2010, 22, 2370–2383. [Google Scholar] [CrossRef] [PubMed]
- Boter, M.; Calleja-Cabrera, J.; Carrera-Castaño, G.; Wagner, G.; Hatzig, S.V.; Abbadi, A.; Snowdon, R.J.; Pernas Ochoa, M.; Oñate-Sánchez, L. An integrative approach to analyze seed germination in Brassica napus. Front. Plant Sci. 2019, 10, 1342. [Google Scholar] [CrossRef]
- Oh, E.; Kim, J.; Park, E.; Kim, J.I.; Kang, C.; Choi, G. PIL5, a phytochrome-interacting basic helix-loop-helix protein, is a key negative regulator of seed germination in Arabidopsis thaliana. Plant Cell 2004, 16, 3045–3058. [Google Scholar] [CrossRef]
- Lorrain, S.; Trevisan, M.; Pradervand, S.; Fankhauser, C. Phytochrome interacting factors 4 and 5 redundantly limit seedling de-etiolation in continuous far-red light. Plant J. 2009, 60, 449–461. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, Q.; Jiang, Y.; Yang, C.; Wang, Q.; Li, L. Shade-induced nuclear localization of PIF7 is regulated by phosphorylation and 14-3-3 proteins in Arabidopsis. Elife 2018, 7, e31636. [Google Scholar] [CrossRef]
- Bhattacharya, J.; Singh, U.K.; Ranjan, A. Interaction of light and temperature signaling at the plant interphase: From cue to stress. In Plant Tolerance to Individual and Concurrent Stresses; Springer: New Delhi, India, 2017; pp. 111–132. [Google Scholar]
- Pagnussat, G.C.; Yu, H.J.; Ngo, Q.A.; Rajani, S.; Mayalagu, S.; Johnson, C.S.; Capron, A.; Xie, L.F.; Ye, D.; Sundaresan, V. Genetic and molecular identification of genes required for female gametophyte development and function in Arabidopsis. Development 2005, 132, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Sahni, S.; Prasad, B.D.; Liu, Q.; Grbic, V.; Sharpe, A.; Singh, S.P.; Krishna, P. Overexpression of the brassinosteroid biosynthetic gene DWF4 in Brassica napus simultaneously increases seed yield and stress tolerance. Sci. Rep. 2016, 6, 28298. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Nakano, T.; Gendron, J.; He, J.; Chen, M.; Vafeados, D.; Yang, Y.; Fujioka, S.; Yoshida, S.; Asami, T.; et al. Nuclear-localized BZR1 mediates brassinosteroid-induced growth and feedback suppression of brassinosteroid biosynthesis. Dev. Cell 2002, 2, 505–513. [Google Scholar] [CrossRef]
- Song, X.; Ma, X.; Li, C.; Hu, J.; Yang, Q.; Wang, T.; Wang, L.; Wang, J.; Guo, D.; Ge, W.; et al. Comprehensive analyses of the BES1 gene family in Brassica napus and examination of their evolutionary pattern in representative species. BMC Genom. 2018, 19, 346. [Google Scholar] [CrossRef] [PubMed]
- Saha, G.; Park, J.I.; Jung, H.J.; Ahmed, N.U.; Kayum, M.A.; Kang, J.G.; Nou, I.S. Molecular characterization of BZR transcription factor family and abiotic stress induced expression profiling in Brassica rapa. Plant Physiol. Biochem. 2015, 92, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Rabiger, D.S.; Drews, G.N. MYB64 and MYB119 are required for cellularization and differentiation during female gametogenesis in Arabidopsis thaliana. PLoS Genet 2013, 9, e1003783. [Google Scholar] [CrossRef]
- Mu, R.L.; Cao, Y.R.; Liu, Y.F.; Lei, G.; Zou, H.F.; Liao, Y.; Wang, H.W.; Zhang, W.K.; Ma, B.; Du, J.Z.; et al. An R2R3-type transcription factor gene AtMYB59 regulates root growth and cell cycle progression in Arabidopsis. Cell Res. 2009, 19, 1291–1304. [Google Scholar] [CrossRef]
- Zhong, R.; Richardson, E.A.; Ye, Z.H. The MYB46 transcription factor is a direct target of SND1 and regulates secondary wall biosynthesis in Arabidopsis. Plant Cell 2007, 19, 2776–2792. [Google Scholar] [CrossRef]
- Ruta, V.; Longo, C.; Lepri, A.; De Angelis, V.; Occhigrossi, S.; Costantino, P.; Vittorioso, P. The DOF Transcription Factors in Seed and Seedling Development. Plants 2020, 9, 218. [Google Scholar] [CrossRef]
- He, L.; Su, C.; Wang, Y.; Wei, Z. ATDOF5.8 protein is the upstream regulator of ANAC069 and is responsive to abiotic stress. Biochimie 2015, 110, 17–24. [Google Scholar] [CrossRef]
- Zou, H.F.; Zhang, Y.Q.; Wei, W.; Chen, H.W.; Song, Q.X.; Liu, Y.F.; Zhao, M.Y.; Wang, F.; Zhang, B.C.; Lin, Q.; et al. The transcription factor AtDOF4. 2 regulates shoot branching and seed coat formation in Arabidopsis. Biochem. J. 2013, 449, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dane, F. NAC (NAM/ATAF/CUC) transcription factors in different stresses and their signaling pathway. Acta Physiol. Plant. 2013, 35, 1397–1408. [Google Scholar] [CrossRef]
- Hegedus, D.; Yu, M.; Baldwin, D.; Gruber, M.; Sharpe, A.; Parkin, I.; Whitwill, S.; Lydiate, D. Molecular characterization of Brassica napus NAC domain transcriptional activators induced in response to biotic and abiotic stress. Plant Mol. Biol. 2003, 53, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Rajavel, A.; Klees, S.; Schlüter, J.S.; Bertram, H.; Lu, K.; Schmitt, A.O.; Gültas, M. Unravelling the complex interplay of transcription factors orchestrating the seed oil content in Brassica napus. Int. J. Mol. Sci. 2021, 21. under revision. [Google Scholar]
- Triska, M.; Solovyev, V.; Baranova, A.; Kel, A.; Tatarinova, T.V. Nucleotide patterns aiding in prediction of eukaryotic promoters. PLoS ONE 2017, 12, e0187243. [Google Scholar] [CrossRef]
- Kanehisa, M. The KEGG database. In Novartis Foundation Symposium; Wiley Online Library: Hoboken, NJ, USA, 2002; pp. 91–100. [Google Scholar]
- Chen, F.; Tholl, D.; D’Auria, J.C.; Farooq, A.; Pichersky, E.; Gershenzon, J. Biosynthesis and emission of terpenoid volatiles from Arabidopsis flowers. Plant Cell 2003, 15, 481–494. [Google Scholar] [CrossRef]
- Xiao, Z.; Li, N.; Wang, S.; Sun, J.; Zhang, L.; Zhang, C.; Yang, H.; Zhao, H.; Yang, B.; Wei, L.; et al. Genome-Wide Identification and Comparative Expression Profile Analysis of the Long-Chain Acyl-CoA synthetase (LACS) Gene Family in Two Different Oil Content Cultivars of Brassica napus. Biochem. Genet. 2019, 57, 781–800. [Google Scholar] [CrossRef]
- Wang, H.W.; Zhang, B.; Hao, Y.J.; Huang, J.; Tian, A.G.; Liao, Y.; Zhang, J.S.; Chen, S.Y. The soybean Dof-type transcription factor genes, GmDof4 and GmDof11, enhance lipid content in the seeds of transgenic Arabidopsis plants. Plant J. 2007, 52, 716–729. [Google Scholar] [CrossRef]
- Su, Y.; Liang, W.; Liu, Z.; Wang, Y.; Zhao, Y.; Ijaz, B.; Hua, J. Overexpression of GhDof1 improved salt and cold tolerance and seed oil content in Gossypium hirsutum. J. Plant Physiol. 2017, 218, 222–234. [Google Scholar] [CrossRef]
- Ni, Y.; Zhang, F.C.; Wang, Y.-C.; Pu, F.; Wang, R.; Chai, Y.-R.; Li, J.-N. Cloning and Functional Analysis of Enoyl-CoA Reductase Gene BnECR from Oilseed Rape (Brassica napus L.). Acta Agron. Sin. 2011, 37, 424–432. [Google Scholar] [CrossRef]
- Puyaubert, J.; Dieryck, W.; Costaglioli, P.; Chevalier, S.; Breton, A.; Lessire, R. Temporal gene expression of 3-ketoacyl-CoA reductase is different in high and in low erucic acid Brassica napus cultivars during seed development. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2005, 1687, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.H.; Van Ha, C.; Nishiyama, R.; Watanabe, Y.; Leyva-González, M.A.; Fujita, Y.; Tran, U.T.; Li, W.; Tanaka, M.; Seki, M.; et al. Arabidopsis type B cytokinin response regulators ARR1, ARR10, and ARR12 negatively regulate plant responses to drought. Proc. Natl. Acad. Sci. USA 2016, 113, 3090–3095. [Google Scholar] [CrossRef] [PubMed]
- Toorchi, M.; Dolati, M.; Adalatzadeh-Aghdam, S. Differentially expressed proteins in canola leaf induced by salt stress-a proteomic approach. Int. J. Biosci. 2014, 5, 433–442. [Google Scholar]
- Zhang, Y.; Liang, W.; Shi, J.; Xu, J.; Zhang, D. MYB 56 Encoding a R2 R 3 MYB Transcription Factor Regulates Seed Size in Arabidopsis thaliana. J. Integr. Plant Biol. 2013, 55, 1166–1178. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Ding, G.; Gai, Z.; Zhang, W.; Han, Y.; Li, W. Changes in the gene expression profile of Arabidopsis thaliana under chromium stress. Ecotoxicol. Environ. Saf. 2020, 193, 110302. [Google Scholar] [CrossRef] [PubMed]
- Lata, C.; Mishra, A.K.; Muthamilarasan, M.; Bonthala, V.S.; Khan, Y.; Prasad, M. Genome-wide investigation and expression profiling of AP2/ERF transcription factor superfamily in foxtail millet (Setaria italica L.). PLoS ONE 2014, 9, e113092. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Feng, L.Y.; Wang, J.H.; Fu, Y.F.; Cai, X.; Wang, C.Q.; Du, J.B.; Yuan, M.; Chen, Y.E.; Xu, P.Z.; et al. Two-factor ANOVA of SSH and RNA-seq analysis reveal development-associated Pi-starvation genes in oilseed rape. Planta 2019, 250, 1073–1088. [Google Scholar] [CrossRef]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. bioRxiv 2014, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Gearing, L.J.; Cumming, H.E.; Chapman, R.; Finkel, A.M.; Woodhouse, I.B.; Luu, K.; Gould, J.A.; Forster, S.C.; Hertzog, P.J. CiiiDER: A tool for predicting and analysing transcription factor binding sites. PLoS ONE 2019, 14, e0215495. [Google Scholar] [CrossRef] [PubMed]
- Meckbach, C.; Tacke, R.; Hua, X.; Waack, S.; Wingender, E.; Gültas, M. PC-TraFF: Identification of potentially collaborating transcription factors using pointwise mutual information. BMC Bioinform. 2015, 16, 400. [Google Scholar] [CrossRef]
- Steuernagel, L.; Meckbach, C.; Heinrich, F.; Zeidler, S.; Schmitt, A.O.; Gültas, M. Computational identification of tissue-specific transcription factor cooperation in ten cattle tissues. PLoS ONE 2019, 14, e0216475. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, R.R.; Chebotarov, D.; Duitama, J.; Smith, S.; De la Hoz, J.F.; Mohiyuddin, M.; Wing, R.A.; McNally, K.L.; Tatarinova, T.; Grigoriev, A.; et al. Structural variants in 3000 rice genomes. Genome Res. 2019, 29, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Shahmuradov, I.A.; Umarov, R.K.; Solovyev, V.V. TSSPlant: A new tool for prediction of plant Pol II promoters. Nucleic Acids Res. 2017, 45, e65. [Google Scholar] [CrossRef]
- Kumari, S.; Ware, D. Genome-wide computational prediction and analysis of core promoter elements across plant monocots and dicots. PLoS ONE 2013, 8, e79011. [Google Scholar] [CrossRef]
- Molina, C.; Grotewold, E. Genome wide analysis of Arabidopsis core promoters. BMC Genom. 2005, 6, 25. [Google Scholar] [CrossRef]
- Wiese, L.; Wangmo, C.; Steuernagel, L.; Schmitt, A.O.; Gültas, M. Construction and visualization of dynamic biological networks: Benchmarking the Neo4J Graph Database. In Proceedings of the International Conference on Data Integration in the Life Sciences, Hannover, Germany, 20–21 November 2018; pp. 33–43. [Google Scholar]
- Blazquez, R.; Wlochowitz, D.; Wolff, A.; Seitz, S.; Wachter, A.; Perera-Bel, J.; Bleckmann, A.; Beißbarth, T.; Salinas, G.; Riemenschneider, M.J.; et al. PI3K: A master regulator of brain metastasis-promoting macrophages/microglia. Glia 2018, 66, 2438–2455. [Google Scholar] [CrossRef]
- Kwon, A.T.; Arenillas, D.J.; Hunt, R.W.; Wasserman, W.W. oPOSSUM-3: Advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3 Genes Genomes Genet. 2012, 2, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Kel, A.E.; Gößling, E.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef] [PubMed]
- Sandelin, A.; Alkema, W.; Engström, P.; Wasserman, W.W.; Lenhard, B. JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004, 32, D91–D94. [Google Scholar] [CrossRef] [PubMed]
- Kursa, M.B.; Rudnicki, W.R. Feature selection with the Boruta package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | No. of DEGs | No. of Up-Regulated DEGs | No. of Down-Regulated DEGs |
|---|---|---|---|
| Flower | 11,442 | 5221 | 6221 |
| Leaf | 3234 | 1486 | 1748 |
| Stem | 4198 | 2510 | 1688 |
| Root | 2318 | 1448 | 870 |
| Cultivar | Oil Quality | Oil Content | Biological Replicates |
|---|---|---|---|
| ZS11 | 00 | high | 2 |
| ZY821 | ++ | low | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klees, S.; Lange, T.M.; Bertram, H.; Rajavel, A.; Schlüter, J.-S.; Lu, K.; Schmitt, A.O.; Gültas, M. In Silico Identification of the Complex Interplay between Regulatory SNPs, Transcription Factors, and Their Related Genes in Brassica napus L. Using Multi-Omics Data. Int. J. Mol. Sci. 2021, 22, 789. https://doi.org/10.3390/ijms22020789
Klees S, Lange TM, Bertram H, Rajavel A, Schlüter J-S, Lu K, Schmitt AO, Gültas M. In Silico Identification of the Complex Interplay between Regulatory SNPs, Transcription Factors, and Their Related Genes in Brassica napus L. Using Multi-Omics Data. International Journal of Molecular Sciences. 2021; 22(2):789. https://doi.org/10.3390/ijms22020789
Chicago/Turabian StyleKlees, Selina, Thomas Martin Lange, Hendrik Bertram, Abirami Rajavel, Johanna-Sophie Schlüter, Kun Lu, Armin Otto Schmitt, and Mehmet Gültas. 2021. "In Silico Identification of the Complex Interplay between Regulatory SNPs, Transcription Factors, and Their Related Genes in Brassica napus L. Using Multi-Omics Data" International Journal of Molecular Sciences 22, no. 2: 789. https://doi.org/10.3390/ijms22020789