
Synthesis of Reactive Sulfur Species in Cultured Vascular Endothelial Cells after Exposure to TGF-β1: Induction of Cystathionine γ-Lyase and Cystathionine β-Synthase Expression Mediated by the ALK5-Smad2/3/4 and ALK5-Smad2/3-ATF4 Pathways

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. TGF-β1 Increases RSS Levels in Vascular Endothelial Cells
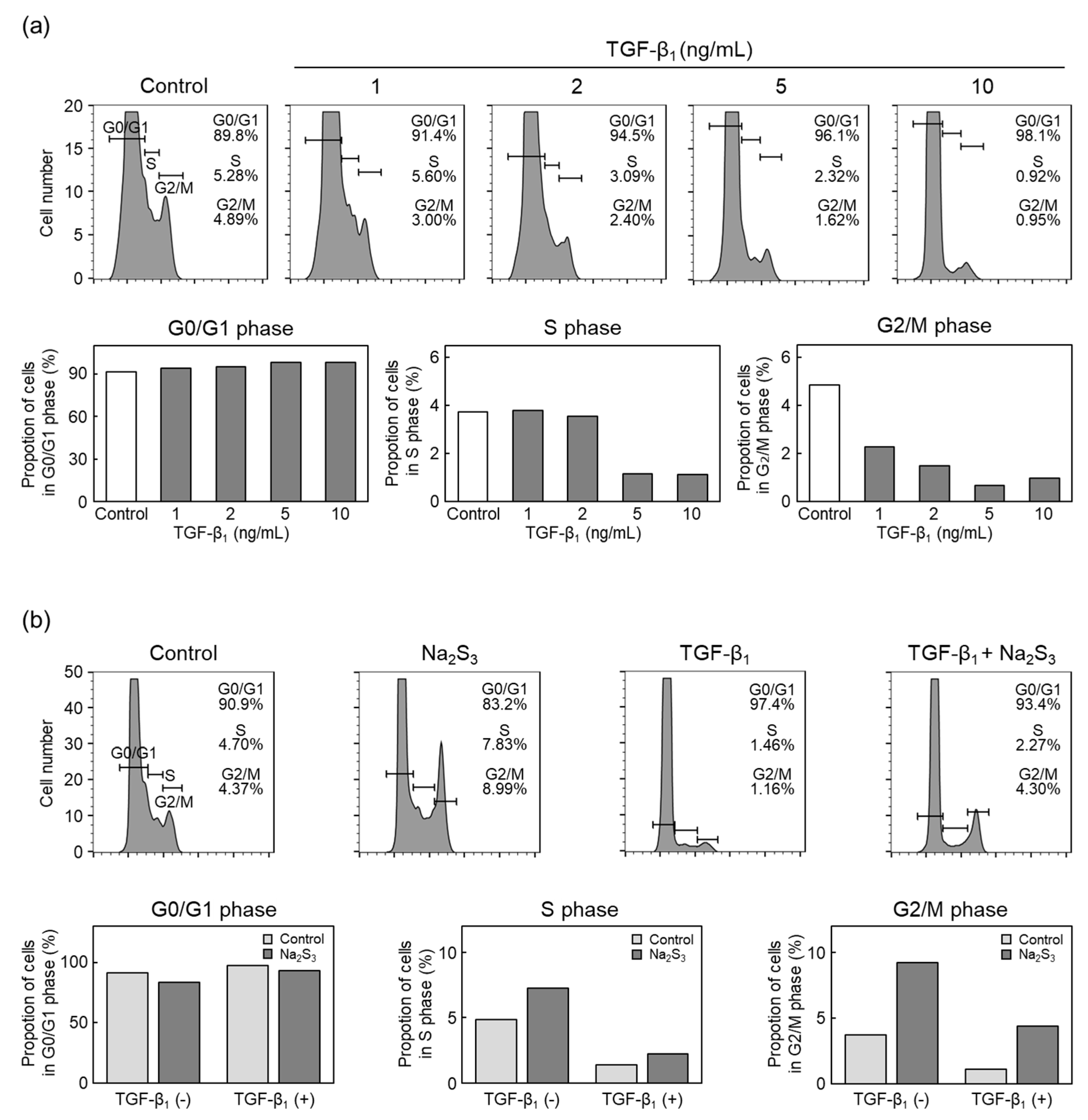
2.2. RSS Modulate TGF-β1 Inhibition of Vascular Endothelial Cell Proliferation
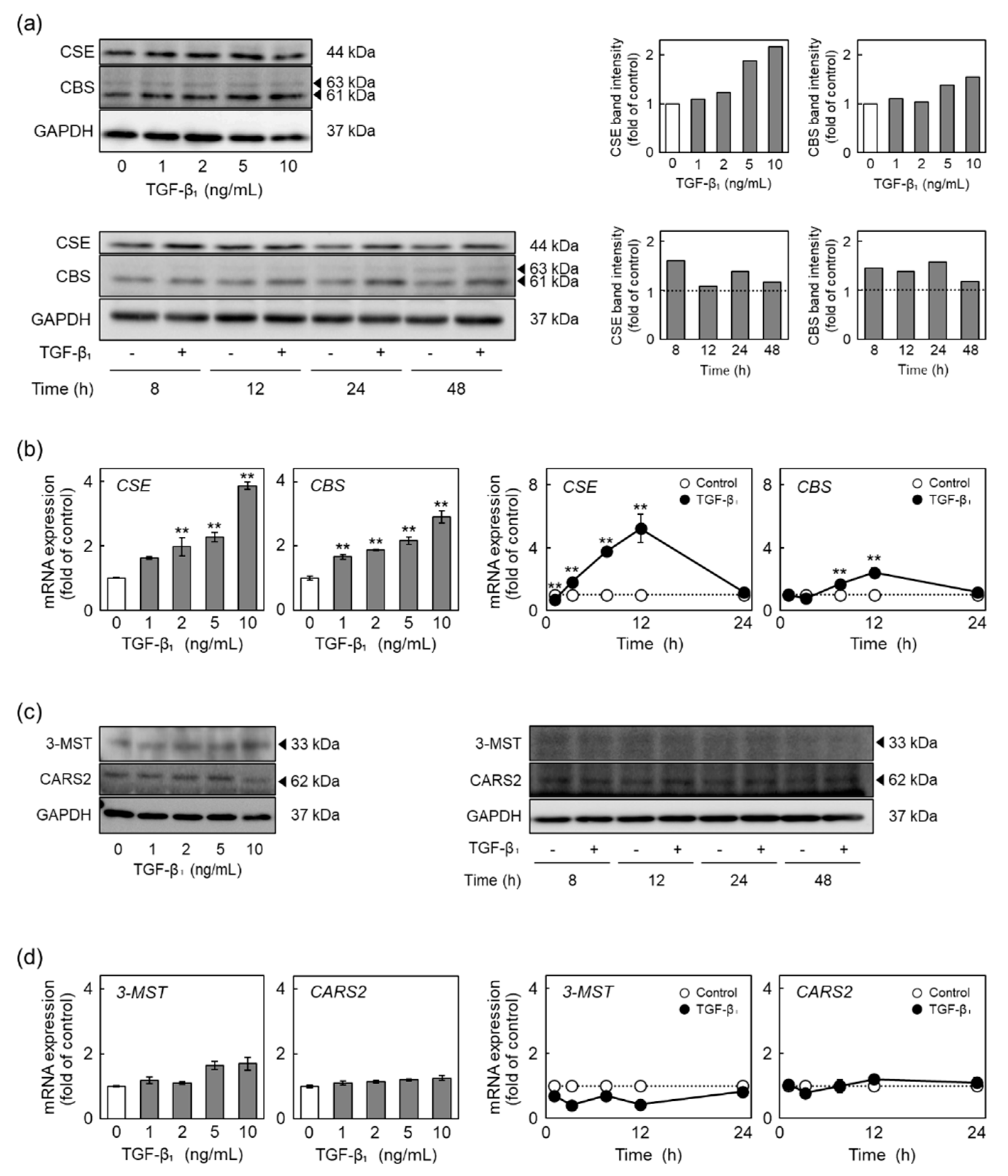
2.3. TGF-β1 Induces the Expression of CSE and Cystathionine β-Synthase (CBS) among RSS-Producing Enzymes
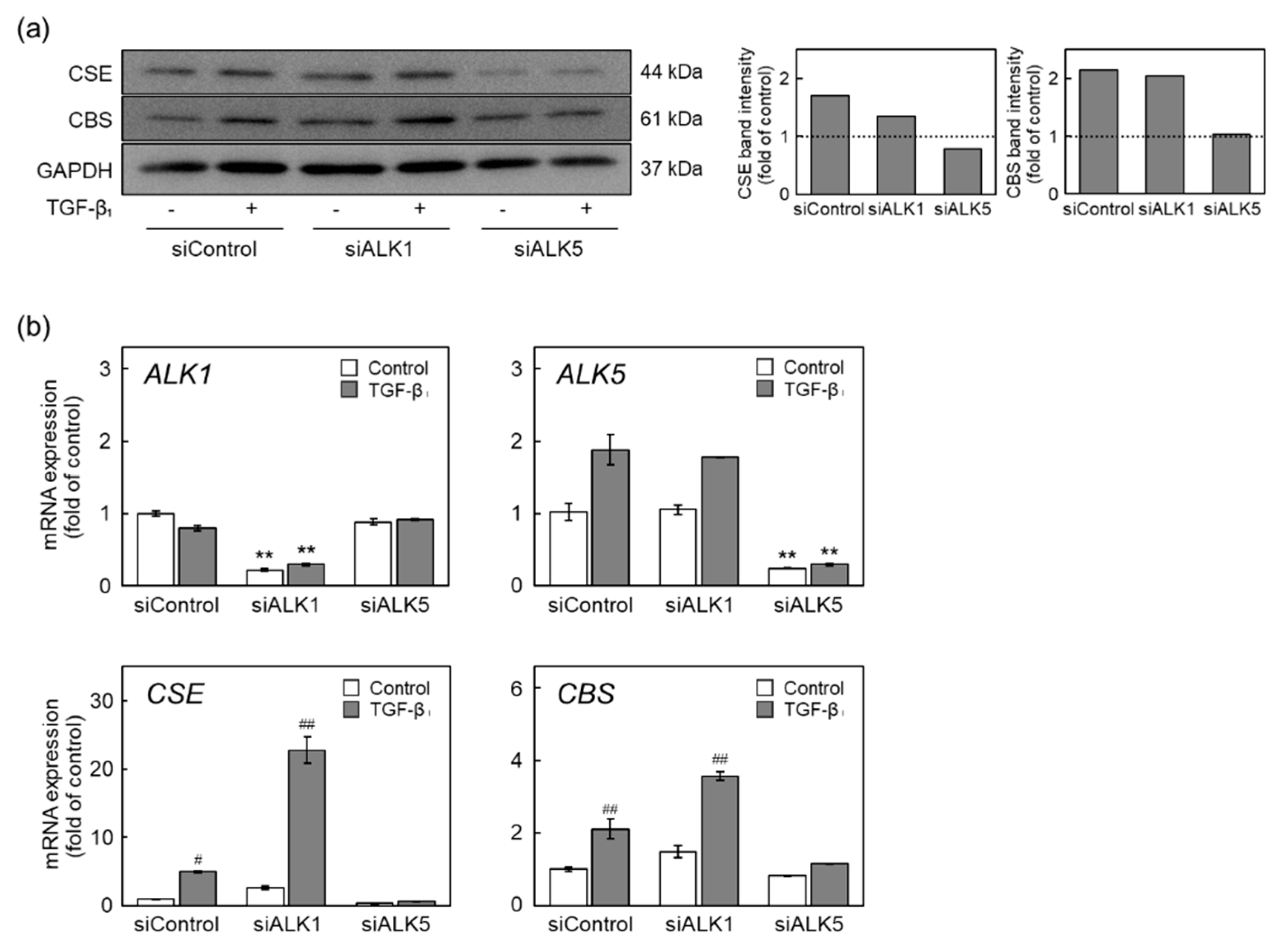
2.4. The ALK5-Smad2/3/4 Pathway Mediates Induction of CSE and CBS Expression by TGF-β1
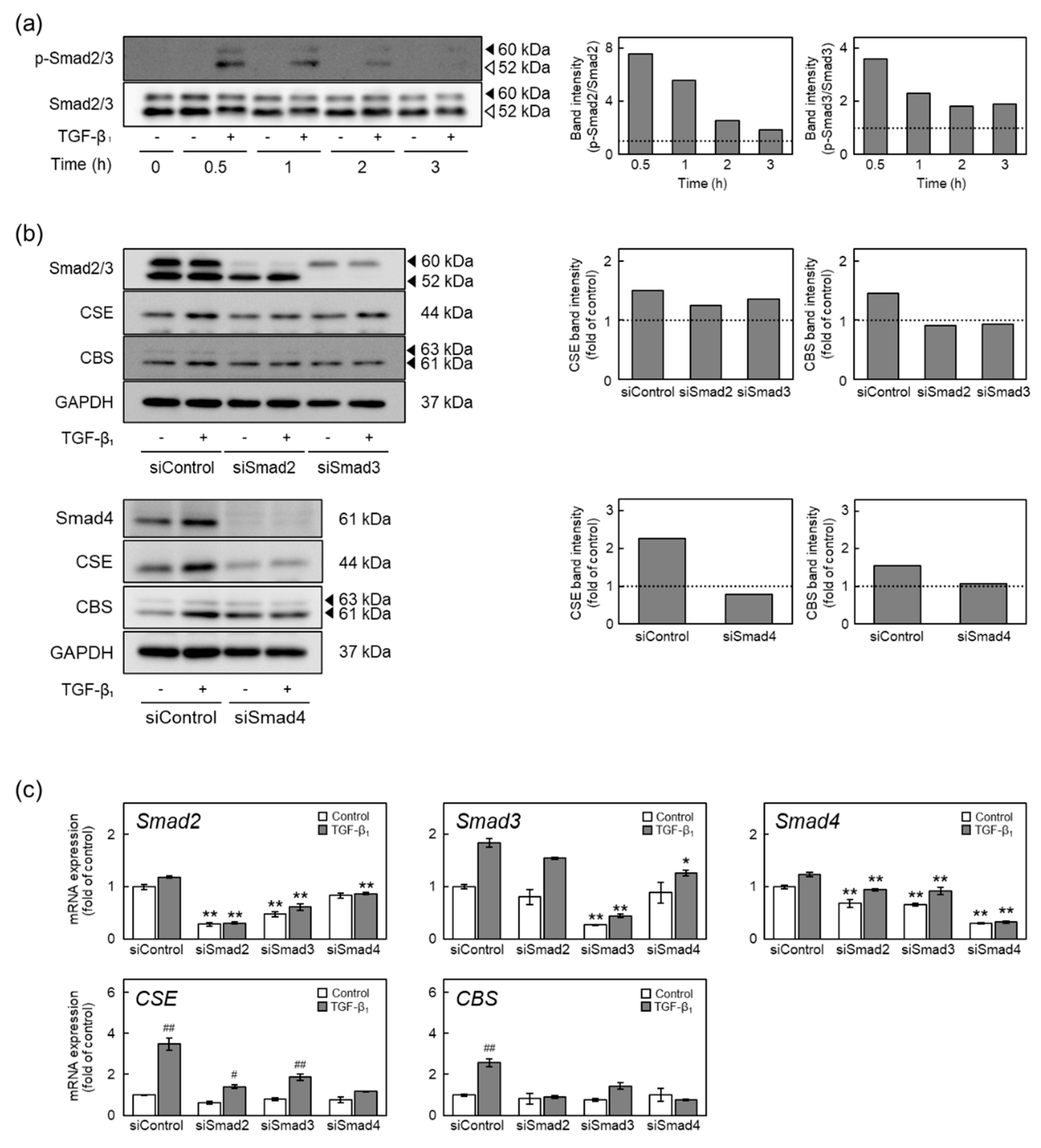
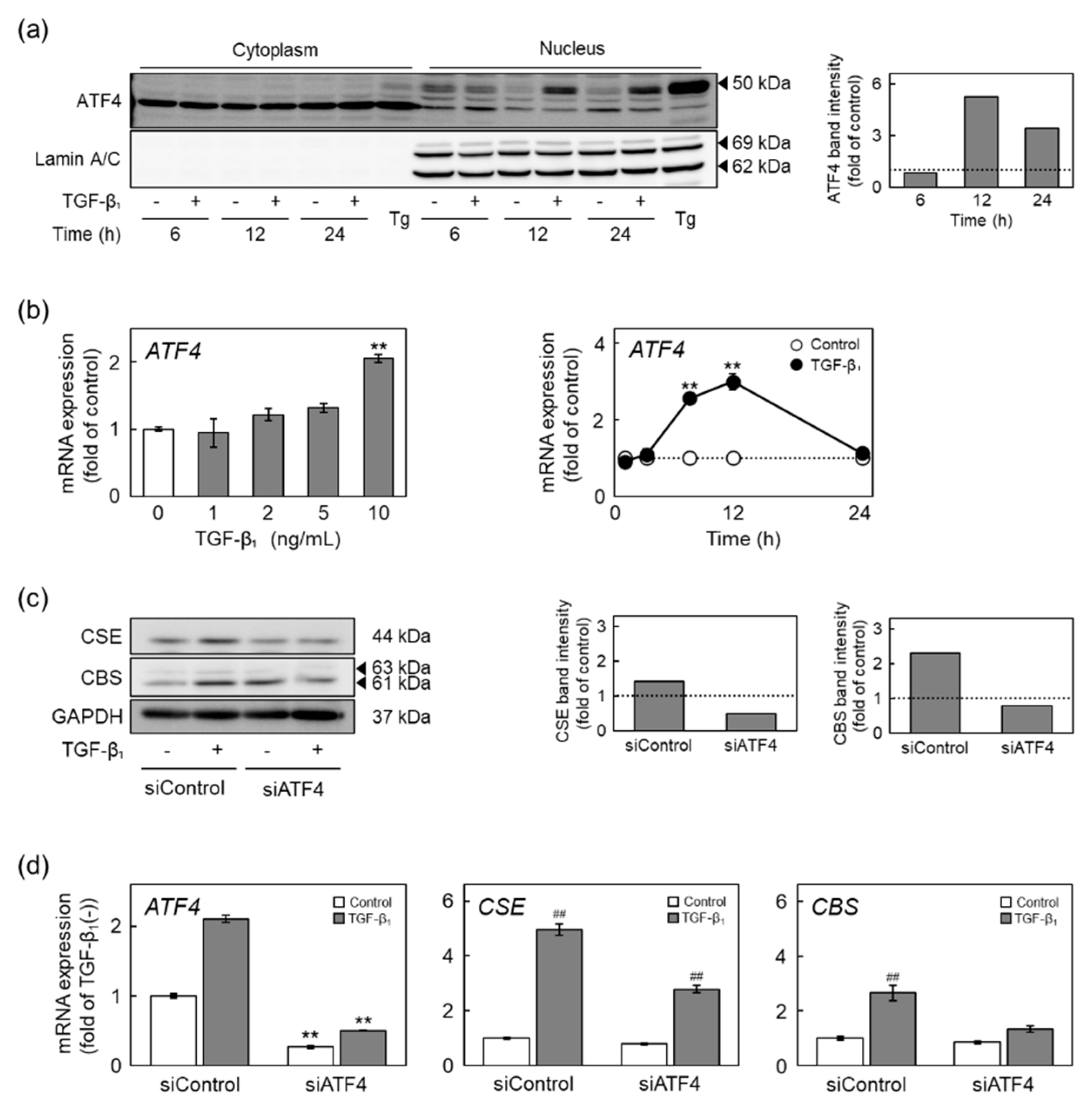
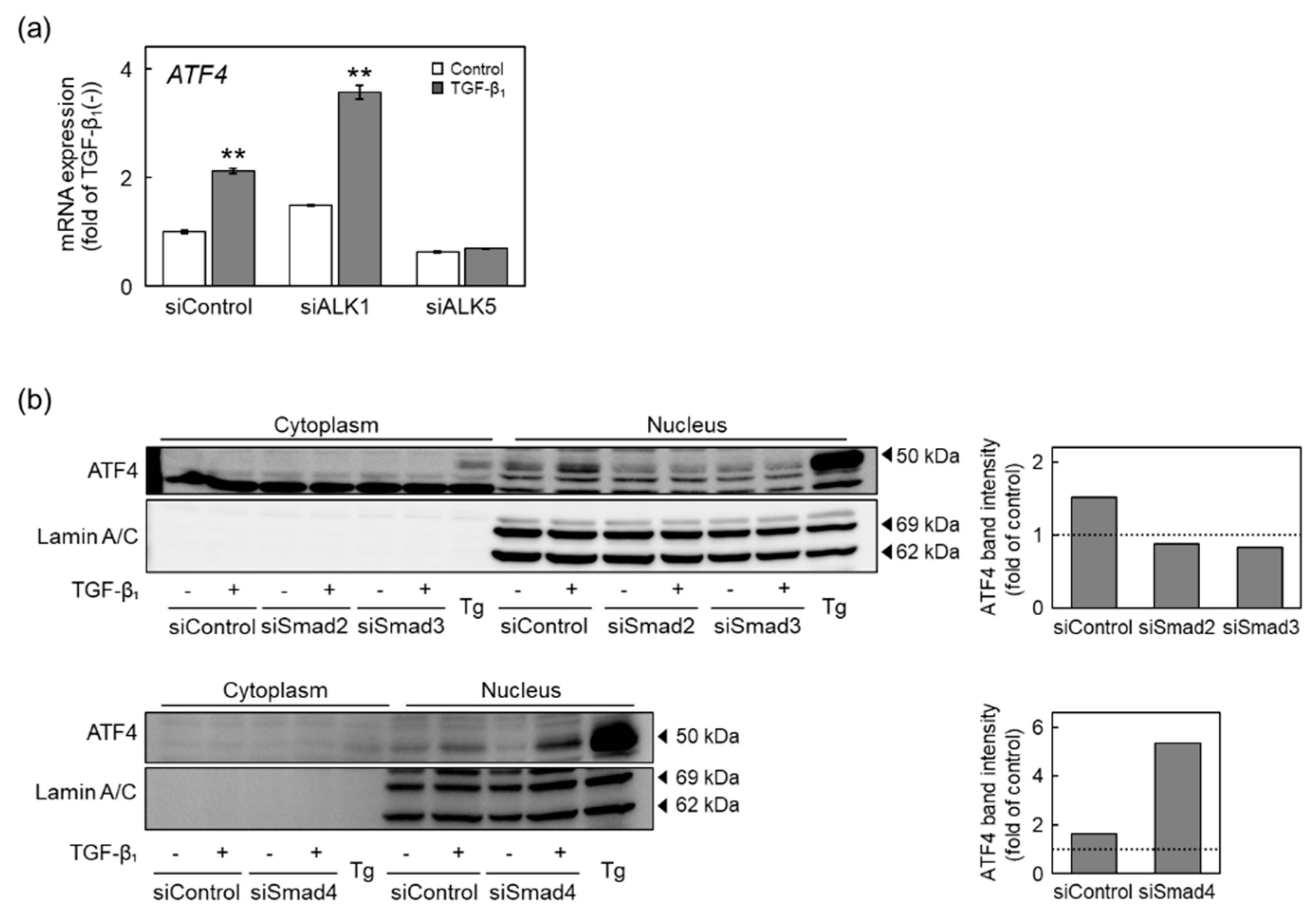
2.5. The ALK5-Smad2/3-ATF4 Pathway Mediates Induction of CSE and CBS Expression by TGF-β1
2.6. The Non-Smad Pathway Does Not Mediate Induction of CSE and CBS Expression by TGF-β1
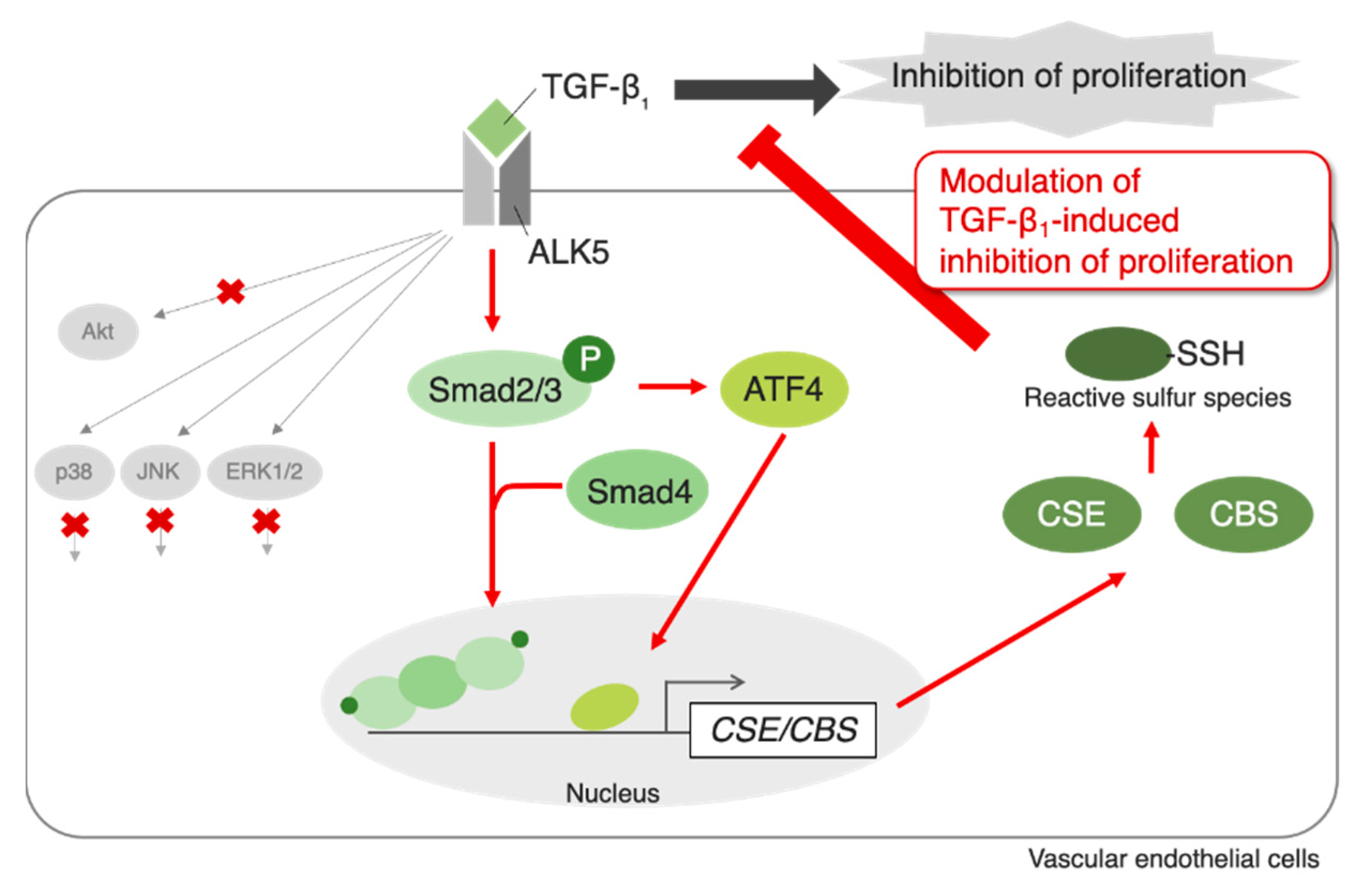
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Measurement of Intracellular RSS Levels
4.3. Cell Cycle Analyses
4.4. siRNA Transfection
4.5. Western Blot Analysis
4.6. Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis
4.7. Statistical Analyses
Author Contributions
Funding
Conflicts of Interest
References
- Weksler, B.B.; Ley, C.W.; Jaffe, E.A. Stimulation of endothelial cell prostacyclin production by thrombin, trypsin, and the ionophore A 23187. J. Clin. Investig. 1978, 62, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Marcum, J.A.; Atha, D.H.; Fritze, L.M.; Nawroth, P.; Stern, D.; Rosenberg, R.D. Cloned bovine aortic endothelial cells synthesize anticoagulantly active heparan sulfate proteoglycan. J. Biol. Chem. 1986, 261, 7507–7517. [Google Scholar] [CrossRef]
- Kinsella, M.G.; Wight, T.N. Isolation and characterization of dermatan sulfate proteoglycans synthesized by cultured bovine aortic endothelial cells. J. Biol. Chem. 1988, 263, 19222–19231. [Google Scholar] [CrossRef]
- Whinna, H.C.; Choi, H.U.; Rosenberg, L.C.; Church, F.C. Interaction of heparin cofactor II with biglycan and decorin. J. Biol. Chem. 1993, 268, 3920–3924. [Google Scholar] [CrossRef]
- Levin, E.G.; Loskutoff, D.J. Cultured bovine endothelial cells produce both urokinase and tissue-type plasminogen activators. J. Cell Biol. 1982, 94, 631–636. [Google Scholar] [CrossRef] [Green Version]
- Ross, R. The pathogenesis of atherosclerosis:a perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Sawdey, M.; Podor, T.J.; Loskutoff, D.J. Regulation of type 1 plasminogen activator inhibitor gene expression in cultured bovine aortic endothelial cells. Induction by transforming growth factor-beta, lipopolysaccharide, and tumor necrosis factor-alpha. J. Biol. Chem. 1989, 264, 10396–10401. [Google Scholar] [CrossRef]
- Heimark, R.L.; Twardzik, D.R.; Schwartz, S.M. Inhibition of endothelial regeneration by type-beta transforming growth factor from platelets. Science 1986, 233, 1078–1080. [Google Scholar] [CrossRef] [PubMed]
- Madri, J.A.; Pratt, B.M.; Tucker, A.M. Phenotypic modulation of endothelial cells by transforming growth factor-beta depends upon the composition and organization of the extracellular matrix. J. Cell Biol. 1988, 106, 1375–1384. [Google Scholar] [CrossRef] [Green Version]
- Kaji, T.; Yamada, A.; Miyajima, S.; Yamamoto, C.; Fujiwara, Y.; Wight, T.N.; Kinsella, M.G. Cell density-dependent regulation of proteoglycan synthesis by transforming growth factor-beta(1) in cultured bovine aortic endothelial cells. J. Biol. Chem. 2000, 275, 1463–1470. [Google Scholar] [CrossRef] [Green Version]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad signaling pathways of the TGF-β family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, E.; Toyama, T.; Shinkai, Y.; Sawa, T.; Akaike, T.; Kumagai, Y. Detoxification of methylmercury by hydrogen sulfide-producing enzyme in mammalian cells. Chem. Res. Toxicol. 2011, 24, 1633–1635. [Google Scholar] [CrossRef] [PubMed]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive Cys persulfides and S-polythiolation regulates oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [Green Version]
- Shinkai, Y.; Masuda, A.; Akiyama, M.; Xian, M.; Kumagai, Y. Cadmium-mediated activation of the HSP90/HSF1 pathway regulated by reactive persulfides/polysulfides. Toxicol. Sci. 2017, 156, 412–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujie, T.; Takahashi, A.; Takahashi, M.; Hara, T.; Soyama, A.; Makino, K.; Takahashi, H.; Yamamoto, C.; Kumagai, Y.; Naka, H.; et al. Transcriptional induction of cystathionine γ-lyase, a reactive sulfur-producing enzyme, by copper diethyldithiocarbamate in cultured vascular endothelial cells. Int. J. Mol. Sci. 2020, 21, 6053. [Google Scholar] [CrossRef]
- Takahashi, M.; Iwai, R.; Takasawa, R.; Nakano, T.; Fujie, T.; Hara, T.; Hara, Y.; Yamamoto, C.; Kaji, T. Sodium trisulfide, a sulfane sulfur donor, stimulates bovine aortic endothelial cell proliferation in culture. J. Toxicol. Sci. 2021, 46, 341–344. [Google Scholar] [CrossRef]
- Takahashi, M.; Kubota, M.; Fujie, T.; Shinkai, Y.; Kumagai, Y.; Nakano, T.; Hara, T.; Yamamoto, C.; Kaji, T. Fibroblast growth factor-2 upregulates the expression of reactive sulfur species by induction of cystathionine γ-lyase via activation of the ERK1/2 signaling pathway in cultured vascular endothelial cells. BPB Rep. 2021, in press. [Google Scholar]
- Fràter-Schröder, M.; Müller, G.; Birchmeier, W.; Böhlen, P. Transforming growth factor-beta inhibits endothelial cell proliferation. Biochem. Biophys. Res. Commun. 1986, 137, 295–302. [Google Scholar] [CrossRef]
- Módis, K.; Asimakopoulou, A.; Coletta, C.; Papapetropoulos, A.; Szabo, C. Oxidative stress suppresses the cellular bioenergetic effect of the 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Biochem. Biophys. Res. Commun. 2013, 33, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistry, R.K.; Murray, T.V.A.; Prysyazhna, O.; Martin, D.; Burgoyne, J.R.; Santos, C.; Eaton, P.; Shah, A.M.; Brewer, A.C. Transcriptional regulation of cystathionine-γ-lyase in endothelial cells by NADPH oxidase 4-dependent signaling. J. Biol. Chem. 2016, 291, 1774–1788. [Google Scholar] [CrossRef] [Green Version]
- Trocha, K.M.; Kip, P.; Tao, M.; MacArthur, M.R.; Treviño-Villarreal, H.; Longchamp, A.; Toussaint, W.; Lambrecht, B.N.; de Vries, M.R.; Quax, P.H.A.; et al. Short-term preoperative protein restriction attenuates vein graft disease via induction of cystathionine γ-lyase. Cardiovasc. Res. 2019, 116, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Tsukuda, A.; Tsuchiya, Y.; Akaik Koike, T.; Watanabe, Y. The active-site cysteine residue of Ca2+/calmodulin-dependent protein kinase I is protected from irreversible modification via generation of polysulfidation. Nitric Oxide 2019, 86, 68–75. [Google Scholar] [CrossRef]
- Doulias, P.T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 2013, 6, rs1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.A.; Wang, T.Y.; Varadharaj, S.; Reyes, L.A.; Hemann, C.; Talukder, M.A.H.; Chen, Y.R.; Druhan, L.J.; Zweier, J.L. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010, 468, 1115–1118. [Google Scholar] [CrossRef] [Green Version]
- Altaany, Z.; Ju, Y.J.; Yang, G.; Wang, R. The coordination of S-sulfhydration, S-nitrosylation, and phosphorylation of endothelial nitric oxide synthase by hydrogen sulfide. Sci. Signal. 2014, 7, ra87. [Google Scholar] [CrossRef] [PubMed]
- Koike, S.; Shibuya, N.; Kimura, H.; Ishii, K.; Ogasawara, Y. Polysulfide promotes neuroblastoma cell differentiation by accelerating calcium influx. Biochem. Biophys. Res. Commun. 2015, 459, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Longen, S.; Richter, F.; Köhler, Y.; Wittig, I.; Beck, K.F.; Pfeilschifter, J. Quantitative persulfide site identification (qPerS-SID) reveals protein targets of H2S releasing donors in mammalian cells. Sci. Rep. 2016, 6, 29808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoji, T.; Hayashi, M.; Sumi, C.; Kusunoki, M.; Uba, T.; Matsuo, Y.; Kimura, H.; Hirota, K. Pharmacological polysulfide suppresses glucose-stimulated insulin secretion in an ATP-sensitive potassium channel-dependent manner. Sci. Rep. 2019, 9, 19377. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.J.; Leng, B.; Wu, Z.Y.; Bian, J.S. Polysulfide and hydrogen sulfide ameliorate cisplatin-induced nephrotoxicity and renal inflammation through Persulfidating STAT3 and IKKβ. Int. J. Mol. Sci. 2020, 21, 7805. [Google Scholar] [CrossRef]
- Sun, H.J.; Xiong, S.P.; Cao, X.; Cao, L.; Zhu, M.Y.; Wu, Z.Y.; Bian, J.S. Polysulfide-mediated sulfhydration of SIRT1 prevents diabetic nephropathy by suppressing phosphorylation and acetylation of p65 NF-κB and STAT3. Redox Biol. 2021, 38, 101813. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Ashino, T.; Fujimori, H.; Numazawa, S. Reactive sulfur species inhibit the migration of PDGF-treated vascular smooth muscle cells by blocking the reactive oxygen species-regulated Akt signaling pathway. Free Radic. Res. 2021, 55, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Leucker, T.M.; Nomura, Y.; Kim, J.H.; Bhatta, A.; Wang, V.; Wecker, A.; Jandu, S.; Santhanam, L.; Berkowitz, D.; Romer, L.; et al. Cystathionine γ-lyase protects vascular endothelium: A role for inhibition of histone deacetylase 6. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H711–H720. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hong, X.; Wang, J.; Jiang, Y.; Zhang, Y.; Chen, J.; Niu, X. Estradiol-17β inhibits homocysteine mediated damage by promoting H2S production via upregulating CBS and CSE expression in human umbilical vein endothelial cells. J. Cell Biochem. 2019, 122, 915–925. [Google Scholar] [CrossRef]
- Song, W.; Ci, H.; Tian, G.; Zhang, Y.; Ge, X. Edoxaban improves venous thrombosis via increasing hydrogen sulfide and homocysteine in rat model. Mol. Med. Rep. 2017, 16, 7706–7714. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Z.; Shi, S.; Gao, F.; Wu, J.; Dong, S.; Zhang, W.; Liu, Y.; Zhong, X. Calcium sensing receptor initiating cystathionine-gamma-lyase/hydrogen sulfide pathway to inhibit platelet activation in hyperhomocysteinemia rat. Exp. Cell Res. 2017, 358, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.P.; Lin, Q.; Tang, C.S.; Liu, X.M. Hydrogen sulfide attenuates epithelial-mesenchymal transition of human alveolar epithelial cells. Pharmacol. Res. 2010, 61, 298–305. [Google Scholar] [CrossRef]
- Lv, M.; Li, Y.; Ji, M.H.; Zhuang, M.; Tang, J.H. Inhibition of invasion and epithelial-mesenchymal transition of human breast cancer cells by hydrogen sulfide through decreased phospho-p38 expression. Mol. Med. Rep. 2014, 10, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Peng, W.; Tao, J.; Lan, Z.; Hei, H.; Tian, L.; Pan, W.; Wang, L.; Zhang, X. Hydrogen sulfide inhibits transforming growth factor-β1-induced EMT via Wnt/catenin pathway. PLoS ONE 2016, 11, e0147018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Zhang, J.; Xie, F.; Tan, W.; Wang, S.; Huang, L.; Tao, L.; Xing, Q.; Yuan, Q. Loss of the protein cystathionine β-synthase during kidney injury promotes renal tubulointerstitial fibrosis. Kidney Blood Press. Res. 2017, 42, 428–443. [Google Scholar] [CrossRef]
- Lebrin, F.; Goumans, M.J.; Jonker, L.; Carvalho, R.L.C.; Valdimarsdottir, G.; Thorikay, M.; Mummery, C.; Arthur, H.M.; ten Dijke, P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004, 23, 4018–4028. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Patel, B.; Harrington, E.O.; Rounds, S. Transforming growth factor-beta1 causes pulmonary microvascular endothelial cell apoptosis via ALK5. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L825–L838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, T.; Yoshida, E.; Fujiwara, Y.; Yamamoto, C.; Kaji, T. Transforming growth factor-β1 modulates the expression of Syndecan-4 in cultured vascular endothelial cells in a biphasic manner. J. Cell. Biochem. 2017, 118, 2009–2017. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Yoshida, E.; Shinkai, Y.; Yamamoto, C.; Fujiwara, Y.; Kumagai, Y.; Kaji, T. Biglycan intensifies ALK5-Smad2/3 signaling by TGF-β1 and downregulates Syndecan-4 in cultured vascular endothelial cells. J. Cell. Biochem. 2017, 118, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Ramachandrarao, S.P.; Siva, S.; Valancius, C.; Zhu, Y.; Mahadev, K.; Toh, I.; Goldstein, B.J.; Woolkalis, M.; Sharma, K. Reactive oxygen species production via NADPH oxidase mediates TGF-beta-induced cytoskeletal alterations in endothelial cells. Am. J. Physiol. Renal Physiol. 2005, 289, F816–F825. [Google Scholar] [CrossRef] [Green Version]
- Saura, M.; Zaragoza, C.; Cao, W.; Bao, C.; Rodríguez-Puyol, M.; Rodríguez-Puyol, D.; Lowenstein, C.J. Smad2 mediates transforming growth factor-beta induction of endothelial nitric oxide synthase expression. Circ. Res. 2002, 91, 806–813. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, M.; Unoki, T.; Shinkai, Y.; Ishii, I.; Ida, T.; Akaike, T.; Yamamoto, M.; Kumagai, Y. Environmental electrophile-mediated toxicity in mice lacking Nrf2, CSE, or both. Environ. Health Perspect. 2019, 127, 67002. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense Strand (5′→3′) | Antisense Strand (5′→3′) |
|---|---|---|
| ALK1 | UUCAUGUCCUCAAAGCUGGGG | CCAGCUUUUGAGGACAUGAAtt |
| ALK5 | UUCAUUUGGCACUCGAUGGUG | CCAUCGAGUGCCAAAUGAAtt |
| Smad2 | UUCAAAACCCUGAUUAACGtt | CGUUAAUCAGGGUUUUGAAtt |
| Smad3 | UGUUUUCGGGGAUGGAAUGtt | CAUUCCAUCCCCGAAAACAtt |
| Smad4 | AAACUCAUCCUGAGUAUGCAU | GCAUACUCAGGAUGAGUUUUG |
| ATF4 | AAUCAAACUCCUUCAAAUCdTdT | GAUUUGAAGGAGUUUGAUUdTdT |
| Negative control | UUCUCCGAACGUGUCACGUtt | ACGUGACACGUUCGGAGAAtt |
| Gene | Sense Primer (5′→3′) | Antisense Primer (5′→3′) |
|---|---|---|
| CSE | TCTCTTGGAGCAGTTCCATCTCCTA | GCAGCCCAGGATAAATAACCTTTTC |
| CBS | GGACTCGGTGCGGAACTACA | GGCAACACGGTCAGCGG |
| 3-MST | GCAGTGGGTGGCTGAGGC | CGATGTCAAAGAAGGCGGC |
| CARS2 | GAGGCGACAGGTACGGCAAG | CAGACTGGCGATGGTGGAAC |
| ALK1 | CAACCACTACTGCTGCTACA | CCATCTCCTTGAGGCTGC |
| ALK5 | GTCTGCTTTGTCTGTATCTCACTCA | TCCTCTTCATTTGGCACTCG |
| Smad2 | CAGAATACCGAAGGCAGACG | TGAGCAACGCACTGAAGG |
| Smad3 | ACTACAGCCATTCCATCC | ATCTGGTGGTCACTGGTCTC |
| Smad4 | CTCCTATTTCTAATCATCCTGCTCC | TCTCAATGGCTTCTGTCCTGTG |
| ATF4 | TGGTCTCAGACAACAGCAAG | AGCTCATCTGGCATGGTTTC |
| B2M | CCATCCAGCGTCCTCCAAAGA | TTCAATCTGGGGTGGATGGAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, M.; Fujie, T.; Nakano, T.; Hara, T.; Shinkai, Y.; Takasawa, R.; Hara, Y.; Kumagai, Y.; Yamamoto, C.; Kaji, T. Synthesis of Reactive Sulfur Species in Cultured Vascular Endothelial Cells after Exposure to TGF-β1: Induction of Cystathionine γ-Lyase and Cystathionine β-Synthase Expression Mediated by the ALK5-Smad2/3/4 and ALK5-Smad2/3-ATF4 Pathways. Int. J. Mol. Sci. 2021, 22, 11762. https://doi.org/10.3390/ijms222111762
Takahashi M, Fujie T, Nakano T, Hara T, Shinkai Y, Takasawa R, Hara Y, Kumagai Y, Yamamoto C, Kaji T. Synthesis of Reactive Sulfur Species in Cultured Vascular Endothelial Cells after Exposure to TGF-β1: Induction of Cystathionine γ-Lyase and Cystathionine β-Synthase Expression Mediated by the ALK5-Smad2/3/4 and ALK5-Smad2/3-ATF4 Pathways. International Journal of Molecular Sciences. 2021; 22(21):11762. https://doi.org/10.3390/ijms222111762
Chicago/Turabian StyleTakahashi, Musubu, Tomoya Fujie, Tsuyoshi Nakano, Takato Hara, Yasuhiro Shinkai, Ryoko Takasawa, Yasushi Hara, Yoshito Kumagai, Chika Yamamoto, and Toshiyuki Kaji. 2021. "Synthesis of Reactive Sulfur Species in Cultured Vascular Endothelial Cells after Exposure to TGF-β1: Induction of Cystathionine γ-Lyase and Cystathionine β-Synthase Expression Mediated by the ALK5-Smad2/3/4 and ALK5-Smad2/3-ATF4 Pathways" International Journal of Molecular Sciences 22, no. 21: 11762. https://doi.org/10.3390/ijms222111762
APA StyleTakahashi, M., Fujie, T., Nakano, T., Hara, T., Shinkai, Y., Takasawa, R., Hara, Y., Kumagai, Y., Yamamoto, C., & Kaji, T. (2021). Synthesis of Reactive Sulfur Species in Cultured Vascular Endothelial Cells after Exposure to TGF-β1: Induction of Cystathionine γ-Lyase and Cystathionine β-Synthase Expression Mediated by the ALK5-Smad2/3/4 and ALK5-Smad2/3-ATF4 Pathways. International Journal of Molecular Sciences, 22(21), 11762. https://doi.org/10.3390/ijms222111762