
Particulate Matter (PM10) Promotes Cell Invasion through Epithelial–Mesenchymal Transition (EMT) by TGF-β Activation in A549 Lung Cells

 ,
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Particulate Matter PM10 Increases the Phosphorylation of SMAD2/3 and the Levels of SMAD4 in A549 Cells
2.2. Particulate Matter PM10 Induce the Expression of TGFB1 and EMT Components in A549 Cells
2.3. Particulate Matter PM10 Increases the Protein Levels of EMT Markers in A549 Cells
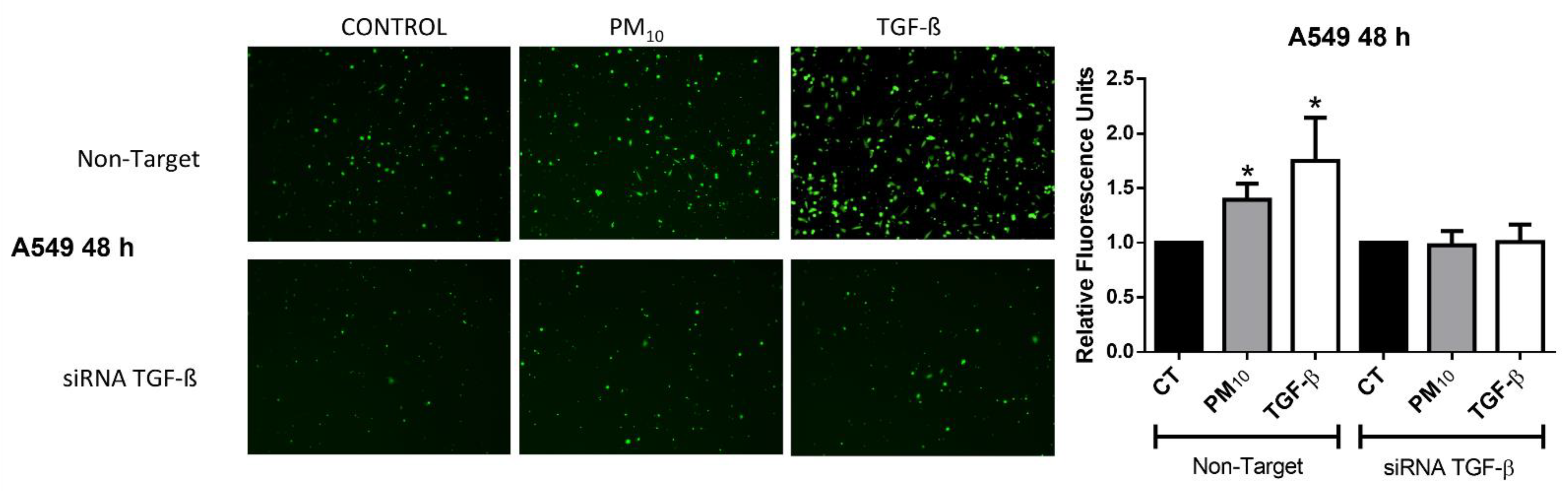
2.4. Particulate Matter PM10 Activates the Invasive Phenotype in A549 Cells
3. Discussion
4. Materials and Methods
4.1. PM10 Sampling
4.2. Lung Epithelial A549 Cell Culture and Exposure
4.3. Knockdown of TGF-β
4.4. Western Blot Analysis
4.5. RNA Isolation and Expression Analysis
4.6. Cell Invasion Assay
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Loomis, D.; Grosse, Y.; Lauby-Secretan, B.; El Ghissassi, F.; Bouvard, V.; Benbrahim-Tallaa, L.; Guha, N.; Baan, R.; Mattock, H.; Straif, K.; et al. The carcinogenicity of outdoor air pollution. Lancet Oncol. 2013, 14, 1262–1263. [Google Scholar] [CrossRef]
- Pope, C.A., 3rd; Burnett, R.T.; Thun, M.J.; Calle, E.E.; Krewski, D.; Ito, K.; Thurston, G.D. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. Jama 2002, 287, 1132–1141. [Google Scholar] [CrossRef] [Green Version]
- de Kok, T.M.; Driece, H.A.; Hogervorst, J.G.; Briede, J.J. Toxicological assessment of ambient and traffic-related particulate matter: A review of recent studies. Mutat. Res. 2006, 613, 103–122. [Google Scholar] [CrossRef]
- Falcon-Rodriguez, C.I.; Osornio-Vargas, A.R.; Sada-Ovalle, I.; Segura-Medina, P. Aeroparticles, Composition, and Lung Diseases. Front. Immunol. 2016, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Turner, M.C.; Andersen, Z.J.; Baccarelli, A.; Diver, W.R.; Gapstur, S.M.; Pope III, C.A.; Prada, D.; Samet, J.; Thurston, G.; Cohen, A. Outdoor air pollution and cancer: An overview of the current evidence and public health recommendations. CA Cancer J. Clin. 2020, 70, 460–479. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- WHO. Ambient Air Pollution: A Global Assessment of Exposure and Burden of Disease; World Health Organization Publications: Geneva, Switzerland, 2016; p. 121. [Google Scholar]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Kourtidis, A.; Lu, R.; Pence, L.J.; Anastasiadis, P.Z. A central role for cadherin signaling in cancer. Exp. Cell Res. 2017, 358, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K.; Katsuno, Y.; Koinuma, D.; Ehata, S.; Morikawa, M. Intracellular and extracellular TGF-β signaling in cancer: Some recent topics. Front. Med. 2018, 12, 387–411. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Zavadil, J.; Böttinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-β on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Shinozaki, M.; Hara, T.; Furuya, T.; Miyazono, K. Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells Devoted Mol. Cell. Mech. 2002, 7, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Chen, Y.G. Regulation of TGF-β receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Vander Ark, A.; Cao, J.; Li, X. TGF-β receptors: In and beyond TGF-β signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Díaz, D. Transforming Growth Factor-β-Induced Cell Plasticity in Liver Fibrosis and Hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [Green Version]
- Guaita, S.; Puig, I.; Franci, C.; Garrido, M.; Dominguez, D.; Batlle, E.; Sancho, E.; Dedhar, S.; De Herreros, A.G.; Baulida, J. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J. Biol. Chem. 2002, 277, 39209–39216. [Google Scholar] [CrossRef] [Green Version]
- Burger, G.A.; Danen, E.H.J.; Beltman, J.B. Deciphering Epithelial-Mesenchymal Transition Regulatory Networks in Cancer through Computational Approaches. Front. Oncol. 2017, 7, 162. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; García De Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Vesuna, F.; van Diest, P.; Chen, J.H.; Raman, V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem. Biophys. Res. Commun. 2008, 367, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Ma, M.; Zhou, W.; Yang, B.; Xiao, C. Inhibition of miR-32 activity promoted EMT induced by PM2.5 exposure through the modulation of the Smad1-mediated signaling pathways in lung cancer cells. Chemosphere 2017, 184, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Libalova, H.; Krckova, S.; Uhlirova, K.; Klema, J.; Ciganek, M.; Rossner, P., Jr.; Sram, R.J.; Vondracek, J.; Machala, M.; Topinka, J. Analysis of gene expression changes in A549 cells induced by organic compounds from respirable air particles. Mutat. Res. 2014, 770, 94–105. [Google Scholar] [CrossRef]
- Wei, H.; Liang, F.; Cheng, W.; Zhou, R.; Wu, X.; Feng, Y.; Wang, Y. The mechanisms for lung cancer risk of PM2.5: Induction of epithelial-mesenchymal transition and cancer stem cell properties in human non-small cell lung cancer cells. Environ. Toxicol. 2017, 32, 2341–2351. [Google Scholar] [CrossRef]
- Chi, Y.; Huang, Q.; Lin, Y.; Ye, G.; Zhu, H.; Dong, S. Epithelial-mesenchymal transition effect of fine particulate matter from the Yangtze River Delta region in China on human bronchial epithelial cells. J. Environ. Sci. 2018, 66, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, C.; Fu, Y.; Guo, L.; Kong, X.; Cai, H. Methylation analysis for multiple gene promoters in non-small cell lung cancers in high indoor air pollution region in China. Bull. Du Cancer 2018, 105, 746–754. [Google Scholar] [CrossRef]
- Yang, B.; Li, X.; Chen, D.; Xiao, C. Effects of fine air particulates on gene expression in non-small-cell lung cancer. Adv. Med. Sci. 2017, 62, 295–301. [Google Scholar] [CrossRef]
- Morales-Barcenas, R.; Chirino, Y.I.; Sanchez-Perez, Y.; Osornio-Vargas, A.R.; Melendez-Zajgla, J.; Rosas, I.; Garcia-Cuellar, C.M. Particulate matter (PM(1)(0)) induces metalloprotease activity and invasion in airway epithelial cells. Toxicol. Lett. 2015, 237, 167–173. [Google Scholar] [CrossRef]
- Yang, B.; Chen, D.; Zhao, H.; Xiao, C. The effects for PM2.5 exposure on non-small-cell lung cancer induced motility and proliferation. SpringerPlus 2016, 5, 2059. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Luo, Q.; Yuan, X.; Chai, L.; Li, D.; Liu, J.; Lv, Z. Atmospheric particulate matter2.5 promotes the migration and invasion of hepatocellular carcinoma cells. Oncol. Lett. 2017, 13, 3445–3450. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Ji, N.; Wang, Z.; Wu, C.; Sun, Z.; Li, Y.; Hu, F.; Wang, Z.; Huang, M.; Zhang, M. Fine Particulate Matter (PM2.5) Promoted the Invasion of Lung Cancer Cells via an ARNT2/PP2A/STAT3/MMP2 Pathway. J. Biomed. Nanotechnol. 2018, 14, 2172–2184. [Google Scholar] [CrossRef] [PubMed]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartis, D.; Mise, N.; Mahida, R.Y.; Eickelberg, O.; Thickett, D.R. Epithelial-mesenchymal transition in lung development and disease: Does it exist and is it important? Thorax 2014, 69, 760–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Nowrin, K.; Sohal, S.S.; Peterson, G.; Patel, R.; Walters, E.H. Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: Fibrosis, remodeling and cancer. Expert Rev. Respir. Med. 2014, 8, 547–559. [Google Scholar] [CrossRef]
- Pain, M.; Bermudez, O.; Lacoste, P.; Royer, P.J.; Botturi, K.; Tissot, A.; Brouard, S.; Eickelberg, O.; Magnan, A. Tissue remodelling in chronic bronchial diseases: From the epithelial to mesenchymal phenotype. Eur. Respir. Rev. Off. J. Eur. Respir. Soc. 2014, 23, 118–130. [Google Scholar] [CrossRef] [Green Version]
- Mittal, V. Epithelial Mesenchymal Transition in Aggressive Lung Cancers. Adv. Exp. Med. Biol. 2016, 890, 37–56. [Google Scholar] [CrossRef]
- Xiao, D.; He, J. Epithelial mesenchymal transition and lung cancer. J. Thorac. Dis. 2010, 2, 154–159. [Google Scholar] [CrossRef]
- Polette, M.; Mestdagt, M.; Bindels, S.; Nawrocki-Raby, B.; Hunziker, W.; Foidart, J.M.; Birembaut, P.; Gilles, C. Beta-catenin and ZO-1: Shuttle molecules involved in tumor invasion-associated epithelial-mesenchymal transition processes. Cells Tissues Organs 2007, 185, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Polette, M.; Nawrocki-Raby, B.; Gilles, C.; Clavel, C.; Birembaut, P. Tumour invasion and matrix metalloproteinases. Crit. Rev. Oncol./Hematol. 2004, 49, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Jung, H.Y.; Fattet, L.; Yang, J. Molecular pathways: Linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 962–968. [Google Scholar] [CrossRef] [Green Version]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.; Daher, N.; Kam, W.; Shafer, M.M.; Ning, Z.; Schauer, J.J.; Sioutas, C. Spatial and temporal variation of chemical composition and mass closure of ambient coarse particulate matter (PM10–2.5) in the Los Angeles area. Atmos. Environ. 2011, 45, 2651–2662. [Google Scholar] [CrossRef]
- Azevedo, R.S.S.; de Sousa, J.R.; Araujo, M.T.F.; Martins Filho, A.J.; de Alcantara, B.N.; Araujo, F.M.C.; Queiroz, M.G.L.; Cruz, A.C.R.; Vasconcelos, B.H.B.; Chiang, J.O.; et al. In situ immune response and mechanisms of cell damage in central nervous system of fatal cases microcephaly by Zika virus. Sci. Rep. 2018, 8, 1. [Google Scholar] [CrossRef]
- Plummer, L.E.; Ham, W.; Kleeman, M.J.; Wexler, A.; Pinkerton, K.E. Influence of Season and Location on Pulmonary Response to California’s San Joaquin Valley Airborne Particulate Matter. J. Toxicol. Environ. Health Part. A 2012, 75, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.Y.; Hoek, G.; Eeftens, M.; de Hoogh, K.; Beelen, R.; Beregszászi, T.; Cesaroni, G.; Cirach, M.; Cyrys, J.; De Nazelle, A.; et al. Spatial variation of PM elemental composition between and within 20 European study areas--Results of the ESCAPE project. Environ. Int. 2015, 84, 181–192. [Google Scholar] [CrossRef]
- Hamra, G.B.; Guha, N.; Cohen, A.; Laden, F.; Raaschou-Nielsen, O.; Samet, J.M.; Vineis, P.; Forastiere, F.; Saldiva, P.; Yorifuji, T.; et al. Outdoor particulate matter exposure and lung cancer: A systematic review and meta-analysis. Environ. Health Perspect. 2014, 122, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Eckel, S.P.; Cockburn, M.; Shu, Y.H.; Deng, H.; Lurmann, F.W.; Liu, L.; Gilliland, F.D. Air pollution affects lung cancer survival. Thorax 2016, 71, 891–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puisieux, A.; Pommier, R.M.; Morel, A.P.; Lavial, F. Cellular Pliancy and the Multistep Process of Tumorigenesis. Cancer Cell 2018, 33, 164–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gos, M.; Miłoszewska, J.; Przybyszewska, M. Epithelial-mesenchymal transition in cancer progression. Postepy Biochem. 2009, 55, 121–128. [Google Scholar] [PubMed]
- Thevenot, P.T.; Saravia, J.; Jin, N.; Giaimo, J.D.; Chustz, R.E.; Mahne, S.; Kelley, M.A.; Hebert, V.Y.; Dellinger, B.; Dugas, T.R.; et al. Radical-containing ultrafine particulate matter initiates epithelial-to-mesenchymal transitions in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2013, 48, 188–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tas, I.; Zhou, R.; Park, S.Y.; Yang, Y.; Gamage, C.D.B.; Son, Y.J.; Paik, M.J.; Kim, H. Inflammatory and tumorigenic effects of environmental pollutants found in particulate matter on lung epithelial cells. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2019, 59, 300–311. [Google Scholar] [CrossRef]
- Cochard, M.; Ledoux, F.; Landkocz, Y. Atmospheric fine particulate matter and epithelial mesenchymal transition in pulmonary cells: State of the art and critical review of the in vitro studies. J. Toxicol. Environ. Health Part B 2020, 23, 293–318. [Google Scholar] [CrossRef]
- Sanchez-Perez, Y.; Chirino, Y.I.; Osornio-Vargas, A.R.; Morales-Barcenas, R.; Gutierrez-Ruiz, C.; Vazquez-Lopez, I.; Garcia-Cuellar, C.M. DNA damage response of A549 cells treated with particulate matter (PM10) of urban air pollutants. Cancer Lett. 2009, 278, 192–200. [Google Scholar] [CrossRef]
- Salcido-Neyoy, M.E.; Sanchez-Perez, Y.; Osornio-Vargas, A.R.; Gonsebatt, M.E.; Melendez-Zajgla, J.; Morales-Barcenas, R.; Petrosyan, P.; Molina-Servin, E.D.; Vega, E.; Manzano-Leon, N.; et al. Induction of c-Jun by air particulate matter (PM10) of Mexico city: Participation of polycyclic aromatic hydrocarbons. Environ. Pollut. 2015, 203, 175–182. [Google Scholar] [CrossRef]
- Chirino, Y.I.; Garcia-Cuellar, C.M.; Garcia-Garcia, C.; Soto-Reyes, E.; Osornio-Vargas, A.R.; Herrera, L.A.; Lopez-Saavedra, A.; Miranda, J.; Quintana-Belmares, R.; Perez, I.R.; et al. Airborne particulate matter in vitro exposure induces cytoskeleton remodeling through activation of the ROCK-MYPT1-MLC pathway in A549 epithelial lung cells. Toxicol. Lett. 2017, 272, 29–37. [Google Scholar] [CrossRef]
- Li, N.; Hao, M.; Phalen, R.F.; Hinds, W.C.; Nel, A.E. Particulate air pollutants and asthma. A paradigm for the role of oxidative stress in PM-induced adverse health effects. Clin. Immunol. 2003, 109, 250–265. [Google Scholar] [CrossRef]
- Ferecatu, I.; Borot, M.C.; Bossard, C.; Leroux, M.; Boggetto, N.; Marano, F.; Baeza-Squiban, A.; Andreau, K. Polycyclic aromatic hydrocarbon components contribute to the mitochondria-antiapoptotic effect of fine particulate matter on human bronchial epithelial cells via the aryl hydrocarbon receptor. Part. Fibre Toxicol. 2010, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Cuellar, C.M.; Santibáñez-Andrade, M.; Chirino, Y.I.; Quintana-Belmares, R.; Morales-Bárcenas, R.; Quezada-Maldonado, E.M.; Sánchez-Pérez, Y. Particulate Matter (PM10) Promotes Cell Invasion through Epithelial–Mesenchymal Transition (EMT) by TGF-β Activation in A549 Lung Cells. Int. J. Mol. Sci. 2021, 22, 12632. https://doi.org/10.3390/ijms222312632
García-Cuellar CM, Santibáñez-Andrade M, Chirino YI, Quintana-Belmares R, Morales-Bárcenas R, Quezada-Maldonado EM, Sánchez-Pérez Y. Particulate Matter (PM10) Promotes Cell Invasion through Epithelial–Mesenchymal Transition (EMT) by TGF-β Activation in A549 Lung Cells. International Journal of Molecular Sciences. 2021; 22(23):12632. https://doi.org/10.3390/ijms222312632
Chicago/Turabian StyleGarcía-Cuellar, Claudia M., Miguel Santibáñez-Andrade, Yolanda I. Chirino, Raúl Quintana-Belmares, Rocío Morales-Bárcenas, Ericka Marel Quezada-Maldonado, and Yesennia Sánchez-Pérez. 2021. "Particulate Matter (PM10) Promotes Cell Invasion through Epithelial–Mesenchymal Transition (EMT) by TGF-β Activation in A549 Lung Cells" International Journal of Molecular Sciences 22, no. 23: 12632. https://doi.org/10.3390/ijms222312632