
Comparing C2=O and C2=S Barbiturates: Different Hydrogen-Bonding Patterns of Thiobarbiturates in Solution and the Solid State

 , and
, and
Abstract
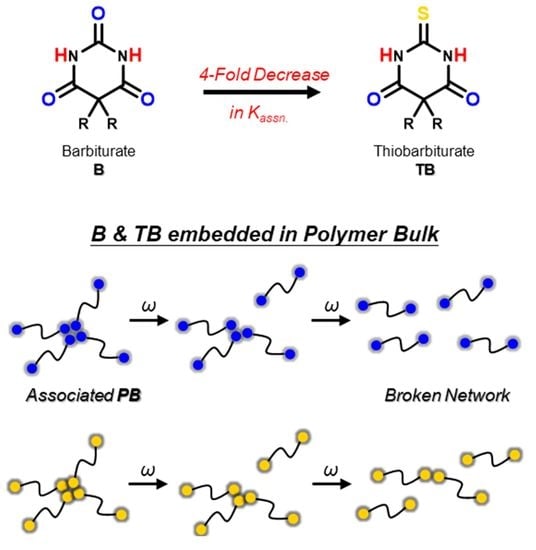
:
1. Introduction
2. Results
Synthesis
3. Discussion
3.1. Hydrogen Bonding of Model Compounds in Solution
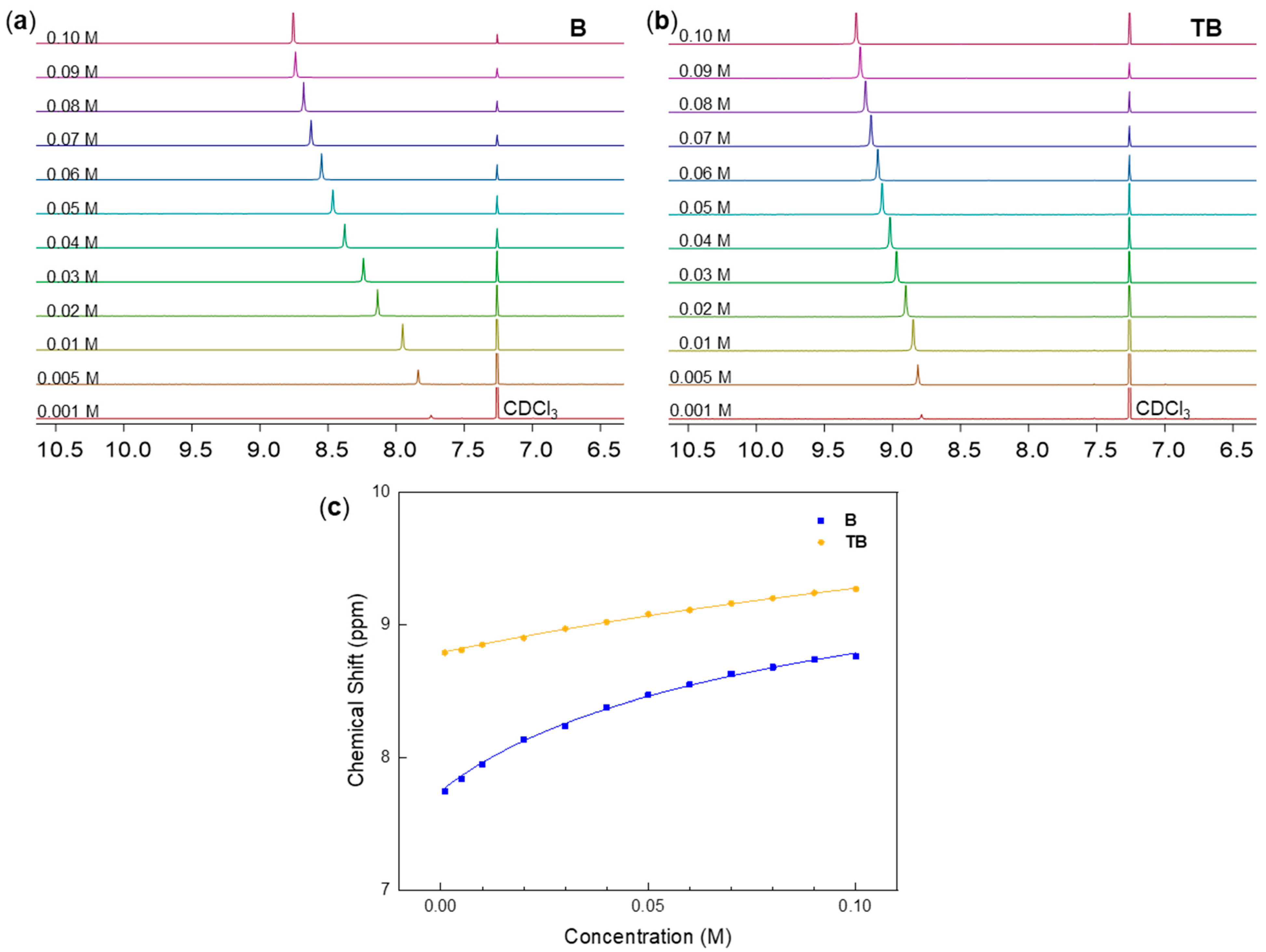
3.1.1. Concentration-Dependent Assembly Studies in Solution
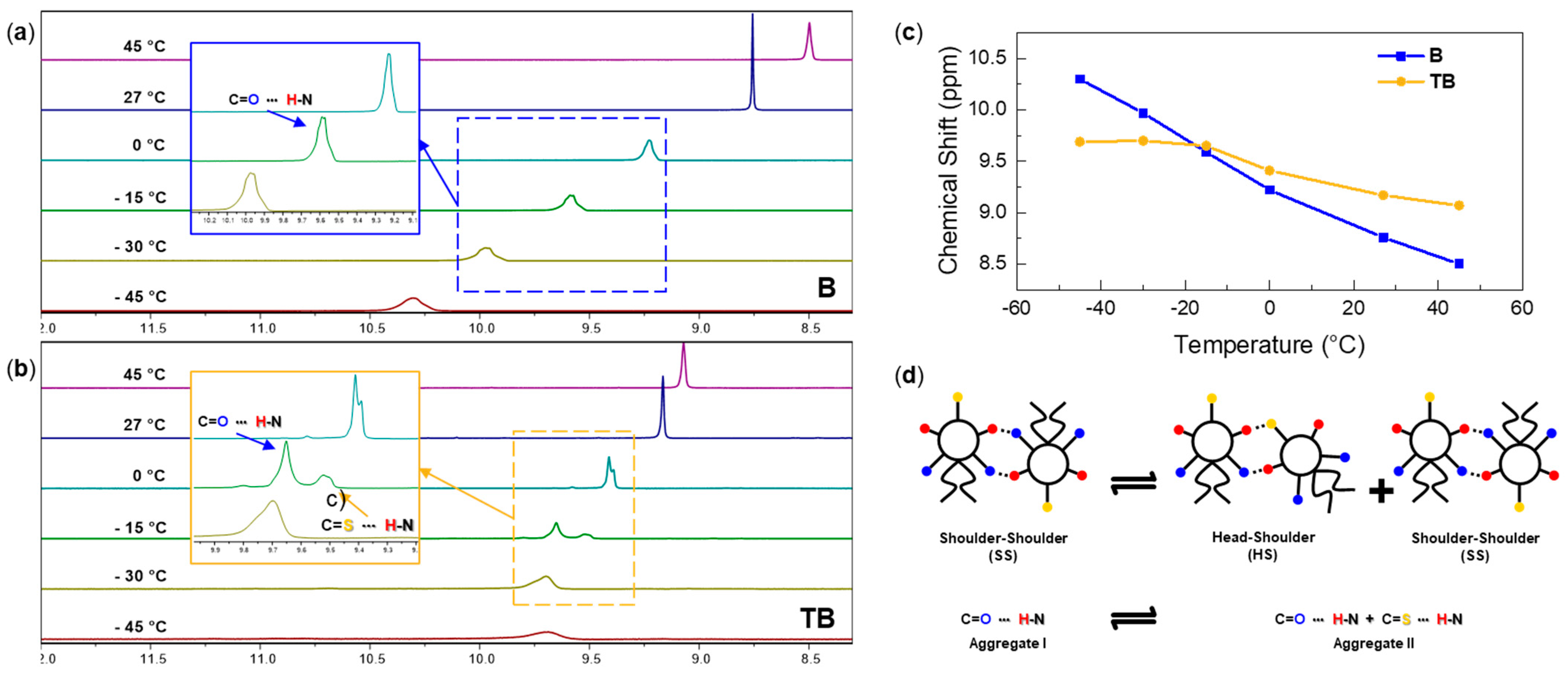
3.1.2. Temperature-Dependent Association Behavior
3.2. Hydrogen Bonding of Model Compounds in the Solid State
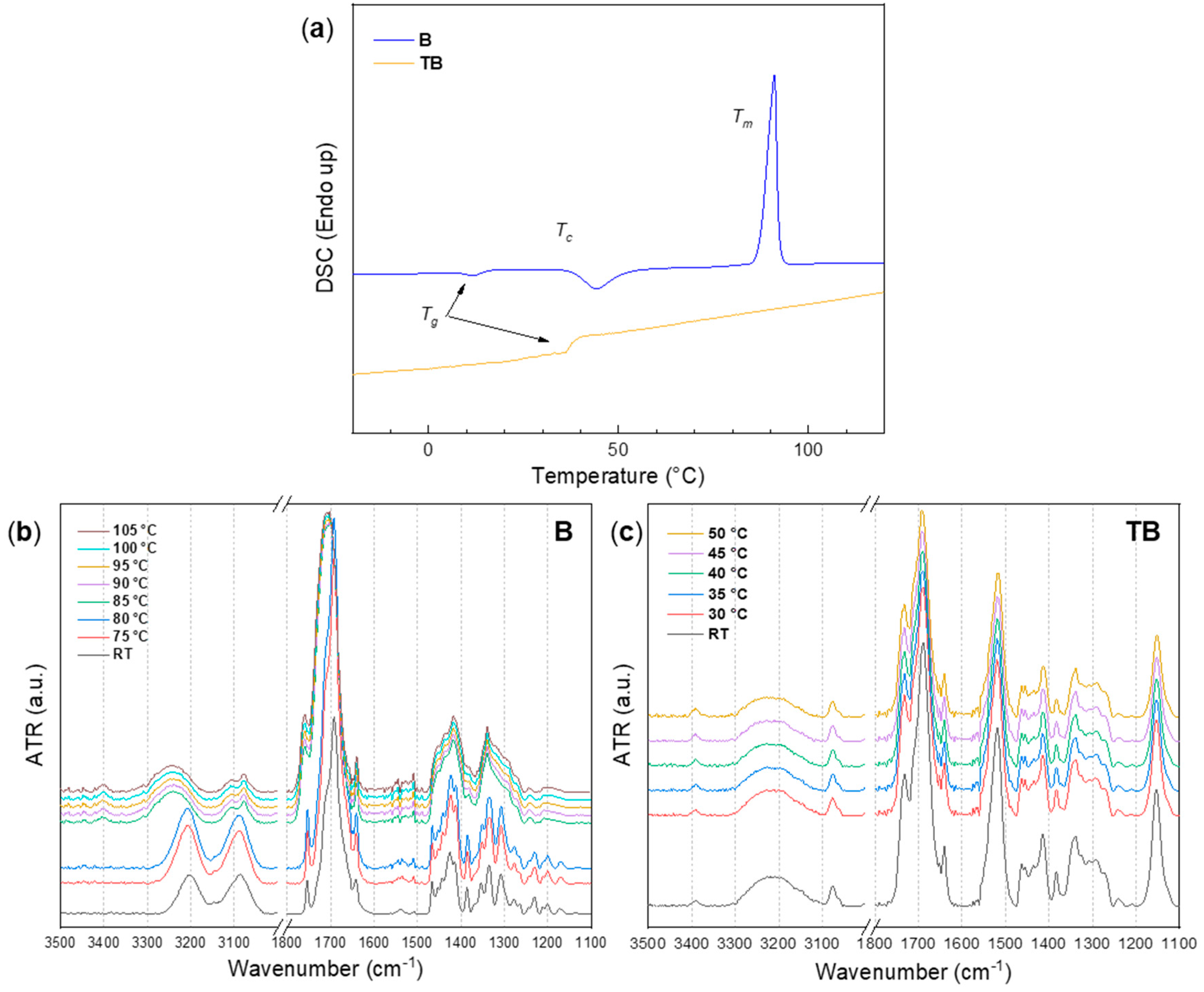
3.2.1. Hydrogen Bonds in Model Compounds B and TB at Elevated Temperatures
3.2.2. Hydrogen Bonds in 2D-Ordered Films
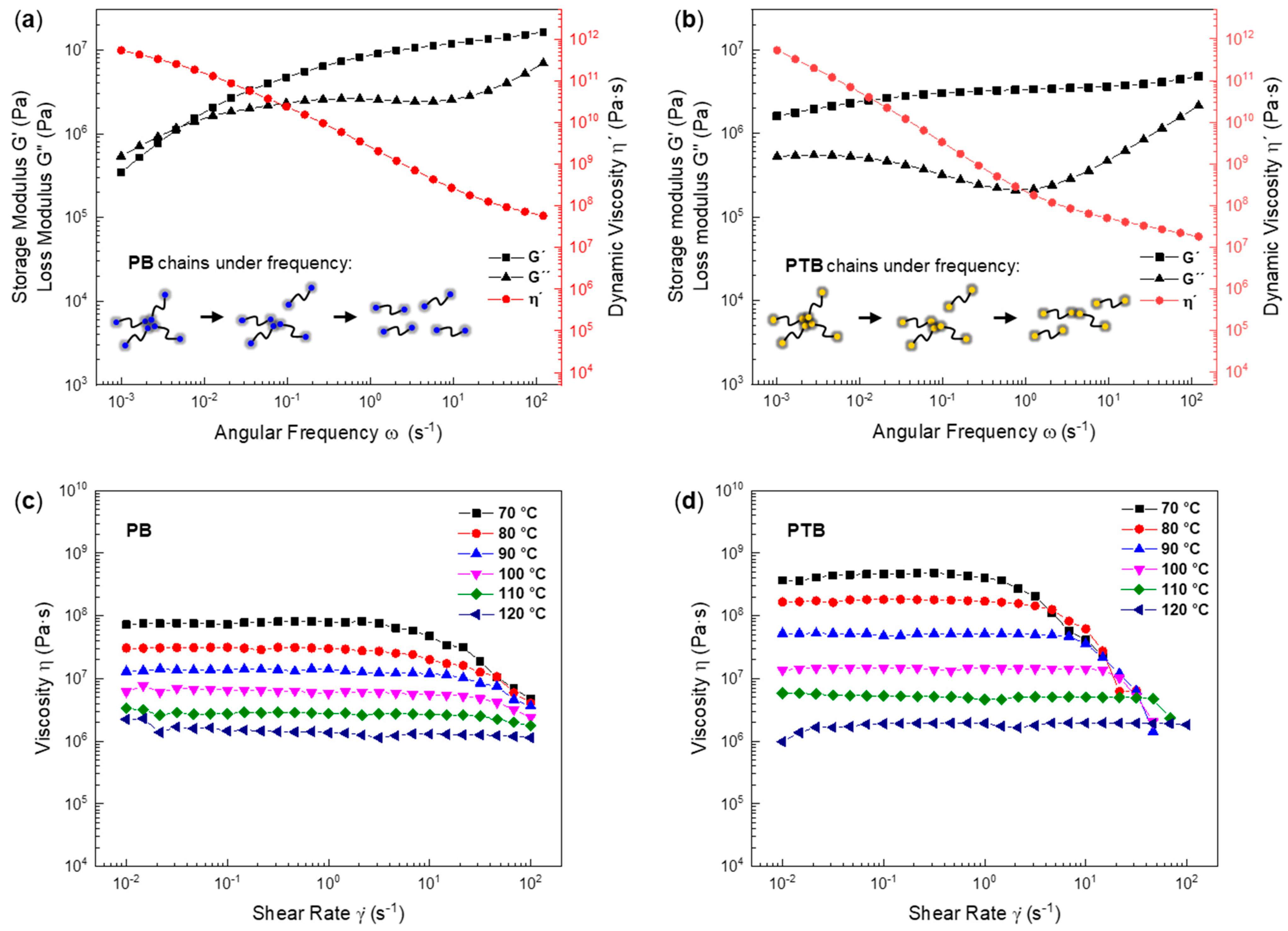
3.3. Hydrogen Bonding of Model Polymers
4. Materials and Methods
4.1. Nuclear Magnetic Resonance Spectroscopy (NMR)
4.2. Differential Scanning Calorimetry (DSC)
4.3. ATR FT-IR Spectroscopy
4.4. Langmuir Isotherm
4.5. Brewster Angle Microscopy
4.6. Rheology
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Weinhold, F.; Klein, R.A. What is a hydrogen bond? Resonance covalency in the supramolecular domain. Chem. Educ. Res. Pract. 2014, 15, 276–285. [Google Scholar] [CrossRef]
- Biswal, H.S.; Chakraborty, S.; Wategaonkar, S. Experimental evidence of O–H—S hydrogen bonding in supersonic jet. J. Chem. Phys. 2008, 129, 184311. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Fu, Y.; Liu, L.; Guo, Q. Hydrogen Bonding Interaction between Ureas or Thioureas and Carbonyl Compounds. Acta Phys.-Chim. Sin. 2007, 23, 1018–1024. [Google Scholar] [CrossRef]
- Lenthall, J.T.; Foster, J.A.; Anderson, K.M.; Probert, M.R.; Howard, J.A.K.; Steed, J.W. Hydrogen bonding interactions with the thiocarbonyl π-system. CrystEngComm 2011, 13, 3202–3212. [Google Scholar] [CrossRef]
- Mundlapati, V.R.; Gautam, S.; Sahoo, D.K.; Ghosh, A.; Biswal, H.S. Thioamide, a Hydrogen Bond Acceptor in Proteins and Nucleic Acids. J. Phys. Chem. Lett. 2017, 8, 4573–4579. [Google Scholar] [CrossRef]
- Chand, A.; Sahoo, D.K.; Rana, A.; Jena, S.; Biswal, H.S. The Prodigious Hydrogen Bonds with Sulfur and Selenium in Molecular Assemblies, Structural Biology, and Functional Materials. Acc. Chem. Res. 2020, 53, 1580–1592. [Google Scholar] [CrossRef] [PubMed]
- Mundlapati, V.R.; Sahoo, D.K.; Ghosh, S.; Purame, U.K.; Pandey, S.; Acharya, R.; Pal, N.; Tiwari, P.; Biswal, H.S. Spectroscopic Evidences for Strong Hydrogen Bonds with Selenomethionine in Proteins. J. Phys. Chem. Lett. 2017, 8, 794–800. [Google Scholar] [CrossRef]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, K.M.; Schreiner, P.R. (Thio)urea Organocatalyst Equilibrium Acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Culik, R.M.; Jo, H.; DeGrado, W.F.; Gai, F. Using Thioamides To Site-Specifically Interrogate the Dynamics of Hydrogen Bond Formation in β-Sheet Folding. J. Am. Chem. Soc. 2012, 134, 8026–8029. [Google Scholar] [CrossRef] [Green Version]
- Stefaniu, C.; Zaffalon, P.-L.; Carmine, A.; Verolet, Q.; Fernandez, S.; Wesolowski, T.A.; Brezesinski, G.; Zumbuehl, A. Rigid Urea and Self-Healing Thiourea Ethanolamine Monolayers. Langmuir 2015, 31, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Hu, R.; Tang, B.Z. Room Temperature One-Step Conversion from Elemental Sulfur to Functional Polythioureas through Catalyst-Free Multicomponent Polymerizations. J. Am. Chem. Soc. 2018, 140, 6156–6163. [Google Scholar] [CrossRef]
- Wu, S.; Luo, M.; Darensbourg, D.J.; Zuo, X. Catalyst-Free Construction of Versatile and Functional CS2-Based Polythioureas: Characteristics from Self-Healing to Heavy Metal Absorption. Macromolecules 2019, 52, 8596–8603. [Google Scholar] [CrossRef]
- Cao, W.; Dai, F.; Hu, R.; Tang, B.Z. Economic Sulfur Conversion to Functional Polythioamides through Catalyst-Free Multicomponent Polymerizations of Sulfur, Acids, and Amines. J. Am. Chem. Soc. 2020, 142, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, Y.; Nan, Y.; Okuro, K.; Aida, T. Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking. Science 2018, 359, 72–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.S.; Choi, Y.-S.; Hwang, J.H.; Lee, J.-H.; Lee, W.; Ahn, S.-k.; Park, S.; Lee, J.-H.; Kim, Y.S.; Kim, D.-G. Weldable and Reprocessable Biomimetic Polymer Networks Based on a Hydrogen Bonding and Dynamic Covalent Thiourea Motif. ACS Appl. Polym. Mater. 2021, 3, 3714–3720. [Google Scholar] [CrossRef]
- Chang, S.K.; Hamilton, A.D. Molecular recognition of biologically interesting substrates: Synthesis of an artificial receptor for barbiturates employing six hydrogen bonds. J. Am. Chem. Soc. 1988, 110, 1318–1319. [Google Scholar] [CrossRef]
- Chang, S.K.; Van Engen, D.; Fan, E.; Hamilton, A.D. Hydrogen bonding and molecular recognition: Synthetic, complexation, and structural studies on barbiturate binding to an artificial receptor. J. Am. Chem. Soc. 1991, 113, 7640–7645. [Google Scholar] [CrossRef]
- McGrath, J.M.; Pluth, M.D. Understanding the Effects of Preorganization, Rigidity, and Steric Interactions in Synthetic Barbiturate Receptors. J. Org. Chem. 2014, 79, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, K.C.; Lehn, J.M.; Kyritsakas, N.; DeCian, A.; Fischer, J. Self-assembly of hydrogen-bonded supramolecular strands from complementary melamine and barbiturate components with chiral selection. New J. Chem. 1998, 22, 123–128. [Google Scholar] [CrossRef]
- Schneider, H.-J.; Juneja, R.K.; Simova, S. Solvent and Structural Effects on Hydrogen Bonds in Some Amides and Barbiturates. An Additive Scheme for the Stability of Corresponding Host-Guest Complexes. Chem. Ber. 1989, 122, 1211–1213. [Google Scholar] [CrossRef]
- Yu, B.-S.; Jo, S.-B.; Kim, C.-K.; Hwang, Y.-S. Interaction between barbiturate and membrane components. Arch. Pharmacal Res. 1990, 13, 246–251. [Google Scholar] [CrossRef]
- Zuccarello, F.; Buemi, G.; Gandolfo, C.; Contino, A. Barbituric and thiobarbituric acids: A conformational and spectroscopic study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2003, 59, 139–151. [Google Scholar] [CrossRef]
- Ramondo, F.; Pieretti, A.; Gontrani, L.; Bencivenni, L. Hydrogen bonding in barbituric and 2-thiobarbituric acids: A theoretical and FT-IR study. Chem. Phys. 2001, 271, 293–308. [Google Scholar] [CrossRef]
- Herbst, F.; Seiffert, S.; Binder, W.H. Dynamic supramolecular poly(isobutylene)s for self-healing materials. Polym. Chem. 2012, 3, 3084–3092. [Google Scholar] [CrossRef]
- Yan, T.; Schröter, K.; Herbst, F.; Binder, W.H.; Thurn-Albrecht, T. What Controls the Structure and the Linear and Nonlinear Rheological Properties of Dense, Dynamic Supramolecular Polymer Networks? Macromolecules 2017, 50, 2973–2985. [Google Scholar] [CrossRef] [Green Version]
- Weck, M.; Fink, R.; Ringsdorf, H. Molecular Recognition via Hydrogen Bonding at the Air−Water Interface: An Isotherm and Fourier Transform Infrared Reflection Spectroscopy Study. Langmuir 1997, 13, 3515–3522. [Google Scholar] [CrossRef]
- Huang, X.; Li, C.; Jiang, S.; Wang, X.; Zhang, B.; Liu, M. Self-Assembled Spiral Nanoarchitecture and Supramolecular Chirality in Langmuir−Blodgett Films of an Achiral Amphiphilic Barbituric Acid. J. Am. Chem. Soc. 2004, 126, 1322–1323. [Google Scholar] [CrossRef]
- Kong, X.; Du, X. In Situ IRRAS Studies of Molecular Recognition of Barbituric Acid Lipids to Melamine at the Air–Water Interface. J. Phys. Chem. B 2011, 115, 13191–13198. [Google Scholar] [CrossRef]
- Sun, M.; Zhou, Y.; Ye, Z.; Li, S.; Dong, A.; Zhang, J. Multifunctional polymer bearing malonylurea groups for the fabrication of coordination complexes and supramolecular assemblies. Eur. Polym. J. 2021, 156, 110616. [Google Scholar] [CrossRef]
- Mahmudov, K.T.; Kopylovich, M.N.; Maharramov, A.M.; Kurbanova, M.M.; Gurbanov, A.V.; Pombeiro, A.J.L. Barbituric acids as a useful tool for the construction of coordination and supramolecular compounds. Coord. Chem. Rev. 2014, 265, 1–37. [Google Scholar] [CrossRef]
- Rupp, H.; Döhler, D.; Hilgeroth, P.; Mahmood, N.; Beiner, M.; Binder, W.H. 3D Printing of Supramolecular Polymers: Impact of Nanoparticles and Phase Separation on Printability. Macromol. Rapid Commun. 2019, 40, 1900467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Mahmood, N.; Beiner, M.; Binder, W.H. Self-Healing Materials from V- and H-Shaped Supramolecular Architectures. Angew. Chem. Int. Ed. 2015, 54, 10188–10192. [Google Scholar] [CrossRef] [PubMed]
- Herbst, F.; Döhler, D.; Michael, P.; Binder, W.H. Self-Healing Polymers via Supramolecular Forces. Macromol. Rapid Commun. 2013, 34, 203–220. [Google Scholar] [CrossRef]
- Chen, S.; Schulz, M.; Lechner, B.-D.; Appiah, C.; Binder, W.H. One-pot synthesis and self-assembly of supramolecular dendritic polymers. Polym. Chem. 2015, 6, 7988–7994. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Ströhl, D.; Binder, W.H. Orthogonal Modification of Polymers via Thio–Bromo “Click” Reaction and Supramolecular Chemistry: An Easy Method Toward Head-to-Tail Self-Assembled Supramolecular Polymers. ACS Macro Lett. 2015, 4, 48–52. [Google Scholar] [CrossRef]
- Mane, S.R.; Sathyan, A.; Shunmugam, R. Barbiturate derived amphiphilic homopolymers: Synthesis, characterization, self-assembly and anticancer drug delivery. Ther. Deliv. 2019, 10, 419–431. [Google Scholar] [CrossRef]
- Mane, S.R.; Rao, N.V.; Shunmugam, R. Reversible pH- and Lipid-Sensitive Vesicles from Amphiphilic Norbornene-Derived Thiobarbiturate Homopolymers. ACS Macro Lett. 2012, 1, 482–488. [Google Scholar] [CrossRef]
- Mane, S.R.; Shunmugam, R. Hierarchical Self-Assembly of Amphiphilic Homopolymer into Unique Superstructures. ACS Macro Lett. 2014, 3, 44–50. [Google Scholar] [CrossRef]
- Bordwell, F.G.; Algrim, D.J.; Harrelson, J.A. The relative ease of removing a proton, a hydrogen atom, or an electron from carboxamides versus thiocarboxamides. J. Am. Chem. Soc. 1988, 110, 5903–5904. [Google Scholar] [CrossRef]
- Alemán, C. On the Ability of Modified Peptide Links to Form Hydrogen Bonds. J. Phys. Chem. A 2001, 105, 6717–6723. [Google Scholar] [CrossRef]
- Abboud, J.L.M.; Roussel, C.; Gentric, E.; Sraidi, K.; Lauransan, J.; Guiheneuf, G.; Kamlet, M.J.; Taft, R.W. Studies on amphiprotic compounds. 3. Hydrogen-bonding basicity of oxygen and sulfur compounds. J. Org. Chem. 1988, 53, 1545–1550. [Google Scholar] [CrossRef]
- Laurence, C.; Berthelot, M.; Le Questel, J.-Y.; El Ghomari, M.J. Hydrogen-bond basicity of thioamides and thioureas. J. Chem. Soc. Perkin Trans. 1995, 2, 2075–2079. [Google Scholar] [CrossRef]
- Allen, F.H.; Bird, C.M.; Rowland, R.S.; Raithby, P.R. Resonance-Induced Hydrogen Bonding at Sulfur Acceptors in R1R2C=S and R1CS2- Systems. Acta Crystallogr. Sect. B 1997, 53, 680–695. [Google Scholar] [CrossRef]
- Sun, C.; Xue, D. IR Spectral Study of Mesoscale Process during Urea Crystallization from Aqueous Solution. Cryst. Growth Des. 2015, 15, 2867–2873. [Google Scholar] [CrossRef]
- Refat, M.S.; El-Korashy, S.A.; Ahmed, A.S. A convenient method for the preparation of barbituric and thiobarbituric acid transition metal complexes. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 71, 1084–1094. [Google Scholar] [CrossRef]
- Srinivasan, K.; Gunasekaran, S.; Krishnan, S. Spectroscopic investigations and structural confirmation studies on thiourea. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 75, 1171–1175. [Google Scholar] [CrossRef] [PubMed]
- Śmiszek-Lindert, W.E.; Chełmecka, E.; Góralczyk, S.; Kaczmarek, M. Vibrational spectroscopic (FT-IR, FT-Raman) studies, Hirshfeld surfaces analysis, and quantum chemical calculations of m-acetotoluidide and m-thioacetotoluidide. J. Mol. Struct. 2017, 1128, 619–628. [Google Scholar] [CrossRef]
- Pegg, R.B.; Shahidi, F.; Jablonski, C.R. Interactions of sulfanilamide and 2-thiobarbituric acid with malonaldehyde: Structure of adducts and implications in determination of oxidative state of nitrite-cured meats. J. Agric. Food Chem. 1992, 40, 1826–1832. [Google Scholar] [CrossRef]
- Laventure, A.; Lauzon, D.; Pellerin, C.; Lebel, O. Triazine-based molecular glasses frustrate the crystallization of barbiturates. CrystEngComm 2019, 21, 1734–1741. [Google Scholar] [CrossRef]
- Hutzler, W.M.; Egert, E.; Bolte, M. One barbiturate and two solvated thiobarbiturates containing the triply hydrogen-bonded ADA/DAD synthon, plus one ansolvate and three solvates of their coformer 2,4-diaminopyrimidine. Acta Crystallogr. Sect. C 2016, 72, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Schröter, K.; Herbst, F.; Binder, W.H.; Thurn-Albrecht, T. Nanostructure and Rheology of Hydrogen-Bonding Telechelic Polymers in the Melt: From Micellar Liquids and Solids to Supramolecular Gels. Macromolecules 2014, 47, 2122–2130. [Google Scholar] [CrossRef]
- Yan, T.; Schröter, K.; Herbst, F.; Binder, W.H.; Thurn-Albrecht, T. Unveiling the molecular mechanism of self-healing in a telechelic, supramolecular polymer network. Sci. Rep. 2016, 6, 32356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, K.E.; Kade, M.J.; Meijer, E.W.; Hawker, C.J.; Kramer, E.J. Model Transient Networks from Strongly Hydrogen-Bonded Polymers. Macromolecules 2009, 42, 9072–9081. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kassn./M−1 | δmax/ppm | ΔGassn./kJ‧mol−1 | kC/s−1 | K−15°C/M−1 | ||
|---|---|---|---|---|---|---|
| B | 4.26 ± 0.40 | 10.67 ± 0.15 | −3.61 ± 2.29 | - | - | - |
| TB | 0.94 ± 0.16 | 12.27 ± 0.46 | 0.15 ± 4.57 | 205.08 | 54.57 | 0.32 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Hilgeroth, P.; Hasan, N.; Ströhl, D.; Kressler, J.; Binder, W.H. Comparing C2=O and C2=S Barbiturates: Different Hydrogen-Bonding Patterns of Thiobarbiturates in Solution and the Solid State. Int. J. Mol. Sci. 2021, 22, 12679. https://doi.org/10.3390/ijms222312679
Li C, Hilgeroth P, Hasan N, Ströhl D, Kressler J, Binder WH. Comparing C2=O and C2=S Barbiturates: Different Hydrogen-Bonding Patterns of Thiobarbiturates in Solution and the Solid State. International Journal of Molecular Sciences. 2021; 22(23):12679. https://doi.org/10.3390/ijms222312679
Chicago/Turabian StyleLi, Chenming, Philipp Hilgeroth, Nazmul Hasan, Dieter Ströhl, Jörg Kressler, and Wolfgang H. Binder. 2021. "Comparing C2=O and C2=S Barbiturates: Different Hydrogen-Bonding Patterns of Thiobarbiturates in Solution and the Solid State" International Journal of Molecular Sciences 22, no. 23: 12679. https://doi.org/10.3390/ijms222312679