
Molecular Mechanisms of Eosinophilic Esophagitis

 ,
,  and
and 
Abstract
:1. Introduction
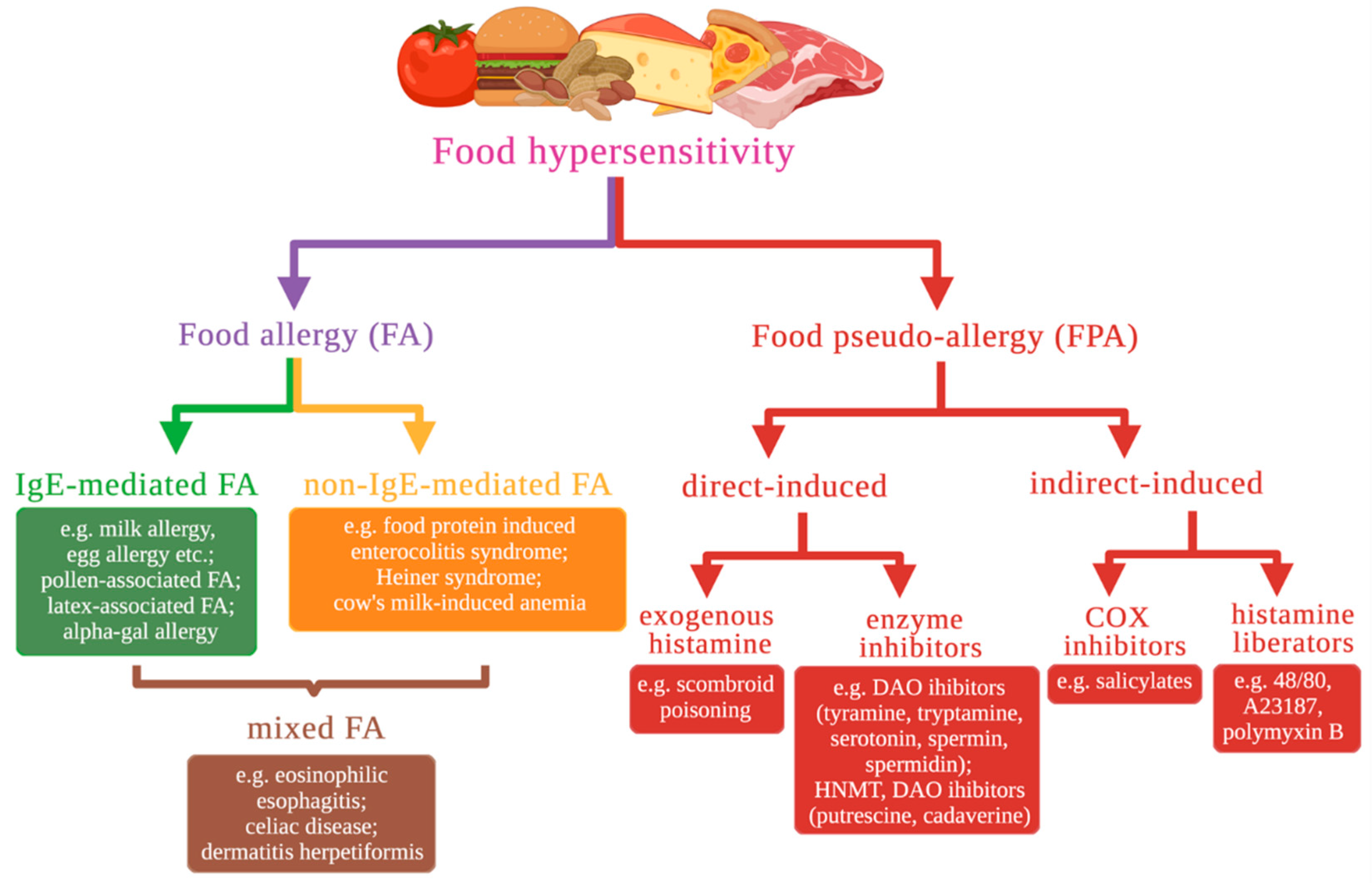
2. Classification and a Brief Description of Food Hypersensitivity Types Based on Molecular Mechanisms
3. Eosinophilic Esophagitis
3.1. The Role of the Eotaxin-3 and IL-13 in the Development of EoE
3.2. Impairment of Esophageal Epithelium Barrier Function
3.3. The Role of the Cadherin 26 in the Development of EoE
3.4. The Role of the Desmosomal Cadherin Desmoglein-1 in the Development of EoE
3.5. Loss of Esophageal Epithelium Differentiation
3.6. The Role of the CAPN14 in the Development of EoE
3.7. The Role of the POSTN in the Development of EoE
3.8. EoE-Associated Risk Genes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, W.; Freeland, D.M.H.; Nadeau, K.C. Food Allergy: Immune Mechanisms, Diagnosis and Immunotherapy. Nat. Rev. Immunol. 2016, 16, 751–765. [Google Scholar] [CrossRef]
- Osborne, N.J.; Koplin, J.J.; Martin, P.E.; Gurrin, L.C.; Lowe, A.J.; Matheson, M.C.; Ponsonby, A.-L.; Wake, M.; Tang, M.L.K.; Dharmage, S.C.; et al. Prevalence of Challenge-Proven IgE-Mediated Food Allergy Using Population-Based Sampling and Predetermined Challenge Criteria in Infants. J. Allergy Clin. Immunol. 2011, 127, 668–676.e1–2. [Google Scholar] [CrossRef] [PubMed]
- Cianferoni, A.; Spergel, J.M. Food Allergy: Review, Classification and Diagnosis. Allergol. Int. 2009, 58, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Johansson, S.G.O.; Hourihane, J.; Bousquet, J.; Bruijnzeel-Koomen, C.; Dreborg, S.; Haahtela, T.; Kowalski, M.L.; Mygind, N.; Ring, J.; Van Cauwenberge, P.; et al. A Revised Nomenclature for Allergy: An EAACI Position Statement from the EAACI Nomenclature Task Force. Allergy 2001, 56, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Ansotegui, I.J.; Melioli, G.; Canonica, G.W.; Caraballo, L.; Villa, E.; Ebisawa, M.; Passalacqua, G.; Savi, E.; Ebo, D.; Gómez, R.M.; et al. IgE Allergy Diagnostics and Other Relevant Tests in Allergy, sa World Allergy Organization Position Paper. World Allergy Organ. J. 2020, 13, 100080. [Google Scholar] [CrossRef]
- Cianferoni, A.; Muraro, A. Food-Induced Anaphylaxis. Immunol. Allergy Clin. N. Am. 2012, 32, 165–195. [Google Scholar] [CrossRef] [Green Version]
- Antonella, C. Non-IgE Mediated Food Allergy. Curr. Pediatr. Rev. 2020, 16, 95–105. [Google Scholar]
- Maintz, L.; Novak, N. Histamine and Histamine Intolerance. Am. J. Clin. Nutr. 2007, 85, 1185–1196. [Google Scholar] [CrossRef]
- Baenkler, H.-W. Salicylate Intolerance. Dtsch. Ärztebl. Int. 2008, 105, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-D.; Cho, K.-S. Samter’s Triad: State of the Art. Clin. Exp. Otorhinolaryngol. 2018, 11, 71–80. [Google Scholar] [CrossRef]
- Zopf, Y.; Hahn, E.G.; Raithel, M.; Baenkler, H.-W.; Silbermann, A. The Differential Diagnosis of Food Intolerance. Dtsch. Ärztebl. Int. 2009, 106, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Alm, P.E. Histamine Liberators and the Mechanisms of Mediator Release. Acta Otolaryngol. 1984, 98, 102–107. [Google Scholar] [CrossRef]
- Lehman, H.K.; Lam, W. Eosinophilic Esophagitis. Pediatr. Clin. N. Am. 2019, 66, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Dellon, E.S.; Liacouras, C.A.; Molina-Infante, J.; Furuta, G.T.; Spergel, J.M.; Zevit, N.; Spechler, S.J.; Attwood, S.E.; Straumann, A.; Aceves, S.S.; et al. Updated International Consensus Diagnostic Criteria for Eosinophilic Esophagitis: Proceedings of the AGREE Conference. Gastroenterology 2018, 155, 1022–1033.e10. [Google Scholar] [CrossRef] [Green Version]
- Hirano, I.; Moy, N.; Heckman, M.G.; Thomas, C.S.; Gonsalves, N.; Achem, S.R. Endoscopic Assessment of the Oesophageal Features of Eosinophilic Oesophagitis: Validation of a Novel Classification and Grading System. Gut 2013, 62, 489–495. [Google Scholar] [CrossRef]
- Gonsalves, N.; Policarpio-Nicolas, M.; Zhang, Q.; Rao, M.S.; Hirano, I. Histopathologic Variability and Endoscopic Correlates in Adults with Eosinophilic Esophagitis. Gastrointest. Endosc. 2006, 64, 313–319. [Google Scholar] [CrossRef]
- Collins, M.H. Histopathologic Features of Eosinophilic Esophagitis. Gastrointest. Endosc. Clin. N. Am. 2008, 18, 59–71. [Google Scholar] [CrossRef]
- Collins, M.H.; Martin, L.J.; Alexander, E.S.; Boyd, J.T.; Sheridan, R.; He, H.; Pentiuk, S.; Putnam, P.E.; Abonia, J.P.; Mukkada, V.A.; et al. Newly Developed and Validated Eosinophilic Esophagitis Histology Scoring System and Evidence That It Outperforms Peak Eosinophil Count for Disease Diagnosis and Monitoring. Dis. Esophagus Off. J. Int. Soc. Dis. Esophagus 2017, 30, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravelli, A.; Villanacci, V.; Cadei, M.; Fuoti, M.; Gennati, G.; Salemme, M. Dilated Intercellular Spaces in Eosinophilic Esophagitis. J. Pediatr. Gastroenterol. Nutr. 2014, 59, 589–593. [Google Scholar] [CrossRef]
- Spergel, J.M.; Brown-Whitehorn, T.F.; Cianferoni, A.; Shuker, M.; Wang, M.-L.; Verma, R.; Liacouras, C.A. Identification of Causative Foods in Children with Eosinophilic Esophagitis Treated with an Elimination Diet. J. Allergy Clin. Immunol. 2012, 130, 461–467.e5. [Google Scholar] [CrossRef]
- Spergel, J.M.; Brown-Whitehorn, T.; Beausoleil, J.L.; Shuker, M.; Liacouras, C.A. Predictive Values for Skin Prick Test and Atopy Patch Test for Eosinophilic Esophagitis. J. Allergy Clin. Immunol. 2007, 119, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Gonsalves, N.; Yang, G.-Y.; Doerfler, B.; Ritz, S.; Ditto, A.M.; Hirano, I. Elimination Diet Effectively Treats Eosinophilic Esophagitis in Adults; Food Reintroduction Identifies Causative Factors. Gastroenterology 2012, 142, 1451–1459.e1. [Google Scholar] [CrossRef] [Green Version]
- Lucendo, A.J. Meta-Analysis-Based Guidance for Dietary Management in Eosinophilic Esophagitis. Curr. Gastroenterol. Rep. 2015, 17, 464. [Google Scholar] [CrossRef]
- Ridolo, E.; Angelis, G.L.D.; Dall’Aglio, P. Eosinophilic Esophagitis after Specific Oral Tolerance Induction for Egg Protein. Ann. Allergy. Asthma. Immunol. 2011, 106, 73–74. [Google Scholar] [CrossRef]
- Miehlke, S.; Alpan, O.; Schröder, S.; Straumann, A. Induction of Eosinophilic Esophagitis by Sublingual Pollen Immunotherapy. Case Rep. Gastroenterol. 2013, 7, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Lucendo, A.J.; Arias, Á.; Tenias, J.M. Relation between Eosinophilic Esophagitis and Oral Immunotherapy for Food Allergy: A Systematic Review with Meta-Analysis. Ann. Allergy. Asthma. Immunol. 2014, 113, 624–629. [Google Scholar] [CrossRef]
- Maggadottir, S.M.; Hill, D.A.; Ruymann, K.; Brown-Whitehorn, T.F.; Cianferoni, A.; Shuker, M.; Wang, M.-L.; Chikwava, K.; Verma, R.; Liacouras, C.A.; et al. Resolution of Acute IgE-Mediated Allergy with Development of Eosinophilic Esophagitis Triggered by the Same Food. J. Allergy Clin. Immunol. 2014, 133, 1487–1489.e1. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Cianferoni, A.; Spergel, J.M.; Aceves, S.; Holbreich, M.; Venter, C.; Rothenberg, M.E.; Terreehorst, I.; Muraro, A.; Lucendo, A.J.; et al. Eosinophilic Esophagitis Is Characterized by a Non-IgE-Mediated Food Hypersensitivity. Allergy 2016, 71, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Loizou, D.; Enav, B.; Komlodi-Pasztor, E.; Hider, P.; Kim-Chang, J.; Noonan, L.; Taber, T.; Kaushal, S.; Limgala, R.; Brown, M.; et al. A Pilot Study of Omalizumab in Eosinophilic Esophagitis. PLoS ONE 2015, 10, e0113483. [Google Scholar] [CrossRef]
- Rocha, R.; Vitor, A.B.; Trindade, E.; Lima, R.; Tavares, M.; Lopes, J.; Dias, J.A. Omalizumab in the Treatment of Eosinophilic Esophagitis and Food Allergy. Eur. J. Pediatr. 2011, 170, 1471. [Google Scholar] [CrossRef] [PubMed]
- Sherrill, J.D.; Kc, K.; Blanchard, C.; Stucke, E.M.; Kemme, K.A.; Collins, M.H.; Abonia, J.P.; Putnam, P.E.; Mukkada, V.A.; Kaul, A.; et al. Analysis and Expansion of the Eosinophilic Esophagitis Transcriptome by RNA Sequencing. Genes Immun. 2014, 15, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, C.; Wang, N.; Stringer, K.F.; Mishra, A.; Fulkerson, P.C.; Abonia, J.P.; Jameson, S.C.; Kirby, C.; Konikoff, M.R.; Collins, M.H.; et al. Eotaxin-3 and a Uniquely Conserved Gene-Expression Profile in Eosinophilic Esophagitis. J. Clin. Investig. 2006, 116, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, C.; Mingler, M.K.; Vicario, M.; Abonia, J.P.; Wu, Y.Y.; Lu, T.X.; Collins, M.H.; Putnam, P.E.; Wells, S.I.; Rothenberg, M.E. IL-13 Involvement in Eosinophilic Esophagitis: Transcriptome Analysis and Reversibility with Glucocorticoids. J. Allergy Clin. Immunol. 2007, 120, 1292–1300. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O.; Nomiyama, H. The Chemokine and Chemokine Receptor Superfamilies and Their Molecular Evolution. Genome Biol. 2006, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, C.; Stucke, E.M.; Rodriguez-Jimenez, B.; Burwinkel, K.; Collins, M.H.; Ahrens, A.; Alexander, E.S.; Butz, B.K.B.; Jameson, S.C.; Kaul, A.; et al. A Striking Local Esophageal Cytokine Expression Profile in Eosinophilic Esophagitis. J. Allergy Clin. Immunol. 2011, 127, 208–217.e7. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, B.; Carlsten, J.; Sabo, E.; Kethu, S.; Meitner, P.; Tavares, R.; Jakate, S.; Mangray, S.; Aswad, B.; Resnick, M.B. Increased Expression of Eotaxin-3 Distinguishes between Eosinophilic Esophagitis and Gastroesophageal Reflux Disease. Hum. Pathol. 2007, 38, 1744–1753. [Google Scholar] [CrossRef]
- Zheng, T.; Zhu, Z.; Wang, Z.; Homer, R.J.; Ma, B.; Riese, R.J.; Chapman, H.A.; Shapiro, S.D.; Elias, J.A. Inducible Targeting of IL-13 to the Adult Lung Causes Matrix Metalloproteinase– and Cathepsin-Dependent Emphysema. J. Clin. Investig. 2000, 106, 1081–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, L.; Fulkerson, P.C.; Finkelman, F.D.; Mingler, M.; Fischetti, C.A.; Blanchard, C.; Rothenberg, M.E. IL-13 Induces Esophageal Remodeling and Gene Expression by an Eosinophil-Independent IL-13Rα2-Inhibited Pathway. J. Immunol. 2010, 185, 660–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, C.; Durual, S.; Estienne, M.; Emami, S.; Vasseur, S.; Cuber, J.-C. Eotaxin-3/CCL26 Gene Expression in Intestinal Epithelial Cells Is up-Regulated by Interleukin-4 and Interleukin-13 via the Signal Transducer and Activator of Transcription 6. Int. J. Biochem. Cell Biol. 2005, 37, 2559–2573. [Google Scholar] [CrossRef] [PubMed]
- Straumann, A.; Bauer, M.; Fischer, B.; Blaser, K.; Simon, H.-U. Idiopathic Eosinophilic Esophagitis Is Associated with a TH2-Type Allergic Inflammatory Response. J. Allergy Clin. Immunol. 2001, 108, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Grendelmeier, P.; Altznauer, F.; Fischer, B.; Bizer, C.; Straumann, A.; Menz, G.; Blaser, K.; Wüthrich, B.; Simon, H.-U. Eosinophils Express Functional IL-13 in Eosinophilic Inflammatory Diseases. J. Immunol. 2002, 169, 1021–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, E.J.; Lu, T.X.; Blanchard, C.; Rothenberg, M.E. Epigenetic Regulation of the IL-13-Induced Human Eotaxin-3 Gene by CREB-Binding Protein-Mediated Histone 3 Acetylation. J. Biol. Chem. 2011, 286, 13193–13204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingras, S.; Simard, J.; Groner, B.; Pfitzner, E. P300/CBP Is Required for Transcriptional Induction by Interleukin-4 and Interacts with Stat6. Nucleic Acids Res. 1999, 27, 2722–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherrill, J.D.; Kc, K.; Wu, D.; Djukic, Z.; Caldwell, J.M.; Stucke, E.M.; Kemme, K.A.; Costello, M.S.; Mingler, M.K.; Blanchard, C.; et al. Desmoglein-1 Regulates Esophageal Epithelial Barrier Function and Immune Responses in Eosinophilic Esophagitis. Mucosal Immunol. 2014, 7, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Page, B.; Vogel, M.; Bussmann, C.; Blanchard, C.; Straumann, A.; Simon, H.-U. Evidence of an Abnormal Epithelial Barrier in Active, Untreated and Corticosteroid-Treated Eosinophilic Esophagitis. Allergy 2018, 73, 239–247. [Google Scholar] [CrossRef]
- Abdulnour-Nakhoul, S.M.; Al-Tawil, Y.; Gyftopoulos, A.A.; Brown, K.L.; Hansen, M.; Butcher, K.F.; Eidelwein, A.P.; Noel, R.A.; Rabon, E.; Posta, A.; et al. Alterations in Junctional Proteins, Inflammatory Mediators and Extracellular Matrix Molecules in Eosinophilic Esophagitis. Clin. Immunol. Orlando Fla 2013, 148, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.; Fernando, S.D.; Biette, K.A.; Hammer, J.A.; Capocelli, K.E.; Kitzenberg, D.A.; Glover, L.E.; Colgan, S.P.; Furuta, G.T.; Masterson, J.C. TGF-Β1 Alters Esophageal Epithelial Barrier Function by Attenuation of Claudin-7 in Eosinophilic Esophagitis. Mucosal Immunol. 2018, 11, 415–426. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, J.M.; Collins, M.H.; Stucke, E.M.; Putnam, P.E.; Franciosi, J.P.; Kushner, J.P.; Abonia, J.P.; Rothenberg, M.E. Histological Eosinophilic Gastritis Is a Systemic Disorder Associated with Blood and Extra-Gastric Eosinophilia, Th2 Immunity, and a Unique Gastric Transcriptome. J. Allergy Clin. Immunol. 2014, 134, 1114–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, J.M.; Collins, M.H.; Kemme, K.A.; Sherrill, J.D.; Wen, T.; Rochman, M.; Stucke, E.M.; Amin, L.; Tai, H.; Putnam, P.E.; et al. Cadherin 26 Is an Alpha Integrin-Binding Epithelial Receptor Regulated during Allergic Inflammation. Mucosal Immunol. 2017, 10, 1190–1201. [Google Scholar] [CrossRef] [Green Version]
- Truong, K.; Ikura, M. The Cadherin Superfamily Database. J. Struct. Funct. Genom. 2002, 2, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, L.; Weis, W.I. Structure and Biochemistry of Cadherins and Catenins. Cold Spring Harb. Perspect. Biol. 2009, 1, a003053. [Google Scholar] [CrossRef]
- Seminario, M.C.; Sterbinsky, S.A.; Bochner, B.S. Beta 1 Integrin-Dependent Binding of Jurkat Cells to Fibronectin Is Regulated by a Serine-Threonine Phosphatase. J. Leukoc. Biol. 1998, 64, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; van Seventer, G.A.; Horgan, K.J.; Shaw, S. Costimulation of Proliferative Responses of Resting CD4+ T Cells by the Interaction of VLA-4 and VLA-5 with Fibronectin or VLA-6 with Laminin. J. Immunol. 1990, 145, 59–67. [Google Scholar]
- Analysis of T Cell Stimulation by Superantigen plus Major Histocompatibility Complex Class II Molecules or by CD3 Monoclonal Antibody: Costimulation by Purified Adhesion Ligands VCAM-1, ICAM-1, but Not ELAM-1. J. Exp. Med. 1991, 174, 901–913. [CrossRef]
- Lehnert, K.; Print, C.G.; Yang, Y.; Krissansen, G.W. MAdCAM-1 Costimulates T Cell Proliferation Exclusively through Integrin Alpha4beta7, Whereas VCAM-1 and CS-1 Peptide Use Alpha4beta1: Evidence for “Remote” Costimulation and Induction of Hyperresponsiveness to B7 Molecules. Eur. J. Immunol. 1998, 28, 3605–3615. [Google Scholar] [CrossRef]
- Berg, R.W.; Yang, Y.; Lehnert, K.; Krissansen, G.W. Mouse M290 Is the Functional Homologue of the Human Mucosal Lymphocyte Integrin HML-1: Antagonism between the Integrin Ligands E-Cadherin and RGD Tripeptide. Immunol. Cell Biol. 1999, 77, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Kawai, K.; Ibusuki, A.; Kanekura, T. Role for E-Cadherin as an Inhibitory Receptor on Epidermal Gammadelta T Cells. J. Immunol. 2011, 186, 6945–6954. [Google Scholar] [CrossRef] [Green Version]
- Gründemann, C.; Bauer, M.; Schweier, O.; von Oppen, N.; Lässing, U.; Saudan, P.; Becker, K.-F.; Karp, K.; Hanke, T.; Bachmann, M.F.; et al. Cutting Edge: Identification of E-Cadherin as a Ligand for the Murine Killer Cell Lectin-like Receptor G1. J. Immunol. 2006, 176, 1311–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.-C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.J.; et al. A Role for IL-25 and IL-33–Driven Type-2 Innate Lymphoid Cells in Atopic Dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef]
- Ito, M.; Maruyama, T.; Saito, N.; Koganei, S.; Yamamoto, K.; Matsumoto, N. Killer Cell Lectin-like Receptor G1 Binds Three Members of the Classical Cadherin Family to Inhibit NK Cell Cytotoxicity. J. Exp. Med. 2006, 203, 289–295. [Google Scholar] [CrossRef]
- Capocelli, K.E.; Fernando, S.D.; Menard-Katcher, C.; Furuta, G.T.; Masterson, J.C.; Wartchow, E.P. Ultrastructural Features of Eosinophilic Oesophagitis: Impact of Treatment on Desmosomes. J. Clin. Pathol. 2015, 68, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, C.; Stucke, E.M.; Burwinkel, K.; Caldwell, J.M.; Collins, M.H.; Ahrens, A.; Buckmeier, B.K.; Jameson, S.C.; Greenberg, A.; Kaul, A.; et al. Coordinate Interaction between IL-13 and Epithelial Differentiation Cluster Genes in Eosinophilic Esophagitis. J. Immunol. 2010, 184, 4033–4041. [Google Scholar] [CrossRef]
- South, A.P.; Ives, J.H.; James, C.H.; Nizetic, D.; Cabral, A.; Mirza, G.; Marenholz, I.; Mischke, D.; Backendorf, C.; Ragoussis, J. Human Epidermal Differentiation Complex in a Single 2.5 Mbp Long Continuum of Overlapping DNA Cloned in Bacteria Integrating Physical and Transcript Maps. J. Investig. Dermatol. 1999, 112, 910–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, A.V.; Harding, C.R. Moisturization and Skin Barrier Function. Dermatol. Ther. 2004, 17 (Suppl. 1), 43–48. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.N.A.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.D.; et al. Common Loss-of-Function Variants of the Epidermal Barrier Protein Filaggrin Are a Major Predisposing Factor for Atopic Dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef]
- Fallon, P.G.; Sasaki, T.; Sandilands, A.; Campbell, L.E.; Saunders, S.P.; Mangan, N.E.; Callanan, J.J.; Kawasaki, H.; Shiohama, A.; Kubo, A.; et al. A Homozygous Frameshift Mutation in the Mouse Flg Gene Facilitates Enhanced Percutaneous Allergen Priming. Nat. Genet. 2009, 41, 602–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rochman, M.; Travers, J.; Miracle, C.E.; Bedard, M.C.; Wen, T.; Azouz, N.P.; Caldwell, J.M.; Kc, K.; Sherrill, J.D.; Davis, B.P.; et al. Profound Loss of Esophageal Tissue Differentiation in Patients with Eosinophilic Esophagitis. J. Allergy Clin. Immunol. 2017, 140, 738–749.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawson, R.; Yang, T.; Newbury, R.O.; Aquino, M.; Doshi, A.; Bell, B.; Broide, D.H.; Dohil, R.; Kurten, R.; Aceves, S.S. TGF-Β1–Induced PAI-1 Contributes to a Profibrotic Network in Patients with Eosinophilic Esophagitis. J. Allergy Clin. Immunol. 2016, 138, 791–800.e4. [Google Scholar] [CrossRef] [Green Version]
- Sevilla, L.M.; Nachat, R.; Groot, K.R.; Klement, J.F.; Uitto, J.; Djian, P.; Määttä, A.; Watt, F.M. Mice Deficient in Involucrin, Envoplakin, and Periplakin Have a Defective Epidermal Barrier. J. Cell Biol. 2007, 179, 1599–1612. [Google Scholar] [CrossRef] [Green Version]
- Eckert, R.L.; Sturniolo, M.T.; Broome, A.-M.; Ruse, M.; Rorke, E.A. Transglutaminase Function in Epidermis. J. Investig. Dermatol. 2005, 124, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleiman, P.M.A.; Wang, M.-L.; Cianferoni, A.; Aceves, S.; Gonsalves, N.; Nadeau, K.; Bredenoord, A.J.; Furuta, G.T.; Spergel, J.M.; Hakonarson, H. GWAS Identifies Four Novel Eosinophilic Esophagitis Loci. Nat. Commun. 2014, 5, 5593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottyan, L.C.; Davis, B.P.; Sherrill, J.D.; Liu, K.; Rochman, M.; Kaufman, K.; Weirauch, M.T.; Vaughn, S.; Lazaro, S.; Rupert, A.M.; et al. Genome-Wide Association Analysis of Eosinophilic Esophagitis Provides Insight into the Tissue Specificity of This Allergic Disease. Nat. Genet. 2014, 46, 895–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litosh, V.A.; Rochman, M.; Rymer, J.K.; Porollo, A.; Kottyan, L.C.; Rothenberg, M.E. Calpain-14 and Its Association with Eosinophilic Esophagitis. J. Allergy Clin. Immunol. 2017, 139, 1762–1771.e7. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.P.; Stucke, E.M.; Khorki, M.E.; Litosh, V.A.; Rymer, J.K.; Rochman, M.; Travers, J.; Kottyan, L.C.; Rothenberg, M.E. Eosinophilic Esophagitis-Linked Calpain 14 Is an IL-13-Induced Protease That Mediates Esophageal Epithelial Barrier Impairment. JCI Insight 2016, 1, e86355. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.E.; Forney, C.; Rochman, M.; Cranert, S.; Habel, J.; Rymer, J.; Lynch, A.; Schroeder, C.; Lee, J.; Sauder, A.; et al. Genetic, Inflammatory, and Epithelial Cell Differentiation Factors Control Expression of Human Calpain-14. G3 Genes Genomes Genet. 2019, 9, 729–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuoka, M.; Shiraishi, H.; Ohta, S.; Suzuki, S.; Arima, K.; Aoki, S.; Toda, S.; Inagaki, N.; Kurihara, Y.; Hayashida, S.; et al. Periostin Promotes Chronic Allergic Inflammation in Response to Th2 Cytokines. J. Clin. Investig. 2012, 122, 2590–2600. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.W.; Annis, D.S.; Mosher, D.F. AMβ2 Integrin–Mediated Adhesion and Motility of IL-5–Stimulated Eosinophils on Periostin. Am. J. Respir. Cell Mol. Biol. 2013, 48, 503–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, M.; Lee, H.-C.; Nakayama, T.; Ziegler, S.F. TSLP Enhances the Function of Helper Type 2 Cells. Eur. J. Immunol. 2011, 41, 1862–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenberg, M.E.; Spergel, J.M.; Sherrill, J.D.; Annaiah, K.; Martin, L.J.; Cianferoni, A.; Gober, L.; Kim, C.; Glessner, J.; Frackelton, E.; et al. Common Variants at 5q22associate with Pediatric Eosinophilic Esophagitis. Nat. Genet. 2010, 42, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Sherrill, J.D.; Gao, P.-S.; Stucke, E.M.; Blanchard, C.; Collins, M.H.; Putnam, P.E.; Franciosi, J.P.; Kushner, J.P.; Abonia, J.P.; Assa’ad, A.H.; et al. Variants of Thymic Stromal Lymphopoietin and Its Receptor Associate with Eosinophilic Esophagitis. J. Allergy Clin. Immunol. 2010, 126, 160–165.e3. [Google Scholar] [CrossRef] [Green Version]
- Blanchard, C.; Mingler, M.; McBride, M.; Putnam, P.; Collins, M.; Chang, G.; Stringer, K.; Abonia, J.; Molkentin, J.; Rothenberg, M. Periostin Facilitates Eosinophil Tissue Infiltration in Allergic Lung and Esophageal Responses. Mucosal Immunol. 2008, 1, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, G.; Arima, K.; Kanaji, T.; Toda, S.; Tanaka, H.; Shoji, S.; McKenzie, A.N.J.; Nagai, H.; Hotokebuchi, T.; Izuhara, K. Periostin: A Novel Component of Subepithelial Fibrosis of Bronchial Asthma Downstream of IL-4 and IL-13 Signals. J. Allergy Clin. Immunol. 2006, 118, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Shao, R. Transduction of a Mesenchyme-Specific Gene Periostin into 293T Cells Induces Cell Invasive Activity through Epithelial-Mesenchymal Transformation. J. Biol. Chem. 2006, 281, 19700–19708. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial-Mesenchymal Transition and Its Implications for Fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Getsios, S.; Simpson, C.L.; Kojima, S.; Harmon, R.; Sheu, L.J.; Dusek, R.L.; Cornwell, M.; Green, K.J. Desmoglein 1–Dependent Suppression of EGFR Signaling Promotes Epidermal Differentiation and Morphogenesis. J. Cell Biol. 2009, 185, 1243–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muir, A.B.; Lim, D.M.; Benitez, A.J.; Modayur Chandramouleeswaran, P.; Lee, A.J.; Ruchelli, E.D.; Spergel, J.M.; Wang, M.-L. Esophageal Epithelial and Mesenchymal Cross-Talk Leads to Features of Epithelial to Mesenchymal Transition in Vitro. Exp. Cell Res. 2013, 319, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagalwalla, A.F.; Akhtar, N.; Woodruff, S.A.; Rea, B.A.; Masterson, J.C.; Mukkada, V.; Parashette, K.R.; Du, J.; Fillon, S.; Protheroe, C.A.; et al. Eosinophilic Esophagitis: Epithelial Mesenchymal Transition Contributes to Esophageal Remodeling and Reverses with Treatment. J. Allergy Clin. Immunol. 2012, 129, 1387–1396.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comeau, M.R.; Ziegler, S.F. The Influence of TSLP on the Allergic Response. Mucosal Immunol. 2010, 3, 138–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, C.C.K.; Rusta-Sallehy, S.; Asher, I.; Heroux, D.; Denburg, J.A. The Effects of Thymic Stromal Lymphopoietin and IL-3 on Human Eosinophil–Basophil Lineage Commitment: Relevance to Atopic Sensitization. Immun. Inflamm. Dis. 2014, 2, 44–55. [Google Scholar] [CrossRef]
- Kottyan, L.C.; Rothenberg, M.E. Genetics of Eosinophilic Esophagitis. Mucosal Immunol. 2017, 10, 580–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakonarson, H.; Grant, S.F.A.; Bradfield, J.P.; Marchand, L.; Kim, C.E.; Glessner, J.T.; Grabs, R.; Casalunovo, T.; Taback, S.P.; Frackelton, E.C.; et al. A Genome-Wide Association Study Identifies KIAA0350 as a Type 1 Diabetes Gene. Nature 2007, 448, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.A.R.; Matheson, M.C.; Tang, C.S.; Granell, R.; Ang, W.; Hui, J.; Kiefer, A.K.; Duffy, D.L.; Baltic, S.; Danoy, P.; et al. Genome-Wide Association Analysis Identifies 11 Risk Variants Associated with the Asthma with Hay Fever Phenotype. J. Allergy Clin. Immunol. 2014, 133, 1564–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, X.; March, M.; Mentch, F.; Nguyen, K.; Glessner, J.; Qu, H.; Liu, Y.; Furuta, G.; Aceves, S.; Gonsalves, N.; et al. A Genome-Wide Association Meta-Analysis Identifies New Eosinophilic Esophagitis Loci. J. Allergy Clin. Immunol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.A.; Vonk, J.M.; Baurecht, H.; Marenholz, I.; Tian, C.; Hoffman, J.D.; Helmer, Q.; Tillander, A.; Ullemar, V.; van Dongen, J.; et al. Shared Genetic Origin of Asthma, Hay Fever and Eczema Elucidates Allergic Disease Biology. Nat. Genet. 2017, 49, 1752–1757. [Google Scholar] [CrossRef]
- Kottyan, L.C.; Maddox, A.; Braxton, J.R.; Stucke, E.M.; Mukkada, V.; Putnam, P.E.; Abonia, J.P.; Chehade, M.; Wood, R.A.; Pesek, R.D.; et al. Genetic Variants at the 16p13 Locus Confer Risk for Eosinophilic Esophagitis. Genes Immun. 2019, 20, 281–292. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EoE Risk Locus | Mapped Gene | Tag SNP | The Strongest SNP Risk Allele | p-Value | OR | Reference |
|---|---|---|---|---|---|---|
| 1p13.3 | LINC02785 SLC25A24 | rs2000260 | A | 7 × 10−7 | 1.32 | [72] |
| 1p32.2 | LINC01767 PLPP3 | rs11206830 | ? | 8 × 10−8 | 2.162 | [72] |
| 1p36.12 | KIF17 | rs2296225 | ? | 1 × 10−7 | 1.626 | [72] |
| 1p36.13 | IFFO2 | rs28530674 | ? | 3 × 10−7 | 1.826 | [72] |
| 2p22.2 | PRKD3 | rs143457389 | A | 3 × 10−16 | 1.77 | [93] |
| 2 × 10−6 | 1.91 | |||||
| 2p23.1 | CAPN14 | rs143457388 | A | 3 × 10−16 | 1.77 | [93] |
| rs149864795 | A | 5 × 10−10 | 2.216 | [71] | ||
| rs77569859 | G | 3 × 10−10 | 1.98 | [72] | ||
| 2q12.1 | TMEM182 | rs887992 | C | 4 × 10−10 | 0.75 | [93] |
| 3q22.1 | CPNE4 | rs554318837 | C | 4 × 10−8 | 2.88 | [93] |
| 3q26.32 | ? | rs6799767 | ? | 4 × 10−7 | 1.49 | [95] |
| 4q21.1 | SHROOM3 | rs13106227 | ? | 4 × 10−6 | 1.52 | [79] |
| rs1986734 | ? | 1 × 10−6 | 1.54 | |||
| 5q14.2 | ? | rs1032757 | T | 2 × 10−6 | 1.96 | [79] |
| 5q22.1 | TSLP | rs3806932 | ? | 3 × 10−9 | 1.85 | [79] |
| rs3806933 | G | 2 × 10−8 | 1.37 | [72] | ||
| TSLP WDR36 | rs252716 | C | 4 × 10−14 | 1.516 | [71] | |
| WDR36 RPS3AP21 | rs1438673 | C | 1 × 10−13 | 1.43 | [93] | |
| 6 × 10−22 | 0.7 | |||||
| 5q23.1 | LINC02214 | rs2055376 | A | 7 × 10−8 | 2.3 | [72] |
| 5q23.2 | LINC02240 | rs4240384 | ? | 2 × 10−7 | 1.4326648 | [95] |
| 5q31.1 | RAD50 | rs2106984 | A | 4 × 10−8 | 1.26 | [93] |
| 6p11.2 | GAPDHP15 | rs9500256 | ? | 5 × 10−6 | 2.04 | [79] |
| 6p21.33 | SNHG32 NEU1 | rs599707 | ? | 3 × 10−9 | 1.6920472 | [95] |
| 6p22.3 | BOLA2P3 | rs1620996 | T | 3 × 10−8 | 0.69 | [93] |
| 7p13 | URGCP-MRPS24 URGCP | rs188483654 | C | 9 × 10−9 | 5.68 | [93] |
| 7p15.1 | JAZF1 | rs11495981 | ? | 9 × 10−7 | 1.308 | [95] |
| 7q22.3 | LARP1BP2 CCDC71L | rs147307036 | A | 1 × 10−8 | 8.04 | [93] |
| 8p23.1 | XKR6 | rs2898261 | C | 5 × 10−8 | 1.35 | [72] |
| 8q22.2 | MATN2 | rs2513845 | T | 7 × 10−9 | 4.18 | [93] |
| ERICH5 | rs13278732 | T | 6 × 10−6 | 1.31 | [79] | |
| 8q24.12 | SNTB1 | rs11989782 | A | 7 × 10−6 | 1.53 | [79] |
| 9p24.1 | JAK2 | rs62541556 | T | 4 × 10−8 | 1.61 | [93] |
| 10p11.21 | CCNY | rs191051238 | C | 4 × 10−8 | 13.2 | [93] |
| 10p12.31 | MIR4675 | rs11819199 | G | 3 × 10−7 | 1.62 | [72] |
| 10q21.1 | PRKG1 | rs185811602 | T | 1 × 10−8 | 6.37 | [93] |
| 10q23.1 | LINC02650 | rs2224865 | G | 9 × 10−6 | 1.44 | [79] |
| 11p15.4 | RHOG STIM1-AS1 | rs147702004 | T | 1 × 10−8 | 1.95 | [93] |
| 11q13.4 | SHANK2 | rs182139615 | T | 1 × 10−9 | 6.62 | [93] |
| 11q13.5 | EMSY | rs61894547 | T | 4 × 10−11 | 2.439 | [71] |
| T | 4 × 10−13 | 1.92 | [93] | |||
| T | 5 × 10−15 | 1.79 | ||||
| EMSY LINC02757 | rs2155219 | A | 4 × 10−7 | 1.37 | [72] | |
| CAPN5 | rs77301713 | ? | 1 × 10−7 | 2.22 | [72] | |
| 11q14.2 | CCDC81 | rs118086209 | C | 2 × 10−7 | 2.19 | [72] |
| 11q21 | FAM76B | rs1939875 | T | 3 × 10−6 | 1.54 | [79] |
| 12q13.3 | STAT6 | rs167769 | T | 2 × 10−7 | 1.351 | [71] |
| T | 2 × 10−6 | 1.36 | [79] | |||
| 13q12.13 | WASF3 GPR12 | rs146034499 | A | 3 × 10−9 | 5.92 | [93] |
| 14q12 | LINC02588 | rs8008716 | G | 7 × 10−8 | 1.712 | [71] |
| 15q13.3 | LINC02352 KLF13 | rs8041227 | G | 6 × 10−12 | 1.52 | [72] |
| 15q22.2 | RORA | rs2279293 | G | 5 × 10−11 | 0.69 | [93] |
| 15q22.33 | SMAD3 | rs56062135 | T | 4 × 10−12 | 1.29 | [93] |
| 16p13.13 | CLEC16A | rs35099084 | C | 3 × 10−9 | 0.71 | [93] |
| T | 2 × 10−12 | 0.72 | ||||
| rs12924112 | ? | 1 × 10−7 | 1.310616 | [95] | ||
| 16q24.1 | MEAK7 | rs371915 | ? | 2 × 10−8 | 1.9 | [79] |
| 17q24.3 | CALM2P1 | rs6501384 | T | 6 × 10−6 | 1.41 | [79] |
| 17q25.3 | CEP295NL TIMP2 | rs3744790 | ? | 8 × 10−7 | 1.54 | [72] |
| 18q12.1 | DSG1 | rs7236477 | G | 7 × 10−6 | 2.22 | [79] |
| 18q12.2 | INO80C GALNT1 | rs534845465 | A | 2 × 10−8 | 5.78 | [93] |
| DCC | rs9956738 | ? | 4 × 10−7 | 2.472 | [72] | |
| 19q13.11 | ANKRD27 | rs3815700 | C | 2 × 10−9 | 1.618 | [71] |
| 21q22.3 | HSF2BP | rs17004598 | C | 1 × 10−7 | 2.57 | [72] |
| 22q11.21 | P2RX6 | rs2075277 | ? | 9 × 10−7 | 1.544 | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhernov, Y.V.; Vysochanskaya, S.O.; Sukhov, V.A.; Zaostrovtseva, O.K.; Gorshenin, D.S.; Sidorova, E.A.; Mitrokhin, O.V. Molecular Mechanisms of Eosinophilic Esophagitis. Int. J. Mol. Sci. 2021, 22, 13183. https://doi.org/10.3390/ijms222413183
Zhernov YV, Vysochanskaya SO, Sukhov VA, Zaostrovtseva OK, Gorshenin DS, Sidorova EA, Mitrokhin OV. Molecular Mechanisms of Eosinophilic Esophagitis. International Journal of Molecular Sciences. 2021; 22(24):13183. https://doi.org/10.3390/ijms222413183
Chicago/Turabian StyleZhernov, Yury V., Sonya O. Vysochanskaya, Vitaly A. Sukhov, Olga K. Zaostrovtseva, Denis S. Gorshenin, Ekaterina A. Sidorova, and Oleg V. Mitrokhin. 2021. "Molecular Mechanisms of Eosinophilic Esophagitis" International Journal of Molecular Sciences 22, no. 24: 13183. https://doi.org/10.3390/ijms222413183